
基于介孔硅的姜黄素⁃siRNA 共递送系统构建及其对巨噬细胞M2 型极化的影响
2021-03-18 02:22李沛汉郎凯宋文
口腔疾病防治 2021年5期
李沛汉, 郎凯, 宋文
1. 军事口腔医学国家重点实验室,口腔疾病国家临床医学研究中心,陕西省口腔医学重点实验室,空军军医大学第三附属医院修复科,陕西 西安(710032); 2. 空军军医大学基础医学院学员二大队七队,陕西 西安(710032)
种植修复目前已逐渐成为口腔牙齿缺失的主要修复方式,其中种植体与骨组织间形成的骨结合是其发挥功能的生物学基础[1]。巨噬细胞是介导固有免疫反应的主体细胞,在外界刺激下可通过向不同方向极化来分泌不同细胞因子,对局部组织的炎症反应发挥调控作用[2]。近年来,巨噬细胞在骨结合形成中的作用越来越受到重视。研究表明,当种植体植入体内后,巨噬细胞首先发生反应,且其极化方向对于生物材料的预后具有显著影响。巨噬细胞的极化可分为M1 和M2 两大类,在种植体植入早期为手术创伤造成的无菌性炎症反应,巨噬细胞向M1 方向极化分泌炎性因子,及时清理局部坏死组织碎片;随着时间推移,巨噬细胞向M2 方向极化,分泌促进组织愈合的生长因子,促进种植体建立骨结合[3]。因此,诱导巨噬细胞M2 方向极化目前已经成为促进种植体骨结合的新策略[4]。
已有研究证实多种刺激均可诱导巨噬细胞向M2 极化,主要包括材料表面的物理形貌刺激[5]、化学基团诱导[6]以及细胞因子刺激[7]等,其中姜黄素(curcumin,CUR)是最为经典的诱导M2 极化的小分子药物之一,其作用的机理主要是通过激活巨噬细胞内过氧化物酶体增殖物激活受体γ(peroxi⁃some proliferator⁃activated receptor γ,PPARγ)诱导M2 极化发生[8],被广泛应用于抗炎治疗中,是极具治疗前景的小分子药物。另一方面,有研究证实miRNA⁃130a⁃3p 为PPARγ内源性抑制分子,采用其反义寡核苷酸(anti⁃sense oligonucleotides,ASO)转染巨噬细胞后可解除miRNA⁃130a⁃3p 对PPARγ的抑制作用,从而促进巨噬细胞M2 极化[9]。因此,笔者推测将ASO 与CUR 共递送至巨噬细胞内,利用ASO 解除miRNA⁃130a⁃3p 对PPARγ的抑制,有利于CUR 与PPARγ受体的结合,高效地诱导巨噬细胞M2 型极化。共递送系统是在一种载体上同时负载两种具有协同作用的药物分子,同时递送至靶细胞后二者可发挥协同作用,进一步增强药物的治疗作用[10]。
基于以上分析,本研究拟采用广泛使用的介孔硅纳米粒(mesoporous silica nanoparticles,MSN)构建共递送系统,利用MSN 内部的规则孔道吸附小分子CUR,在外层利用聚乙烯亚胺(polyethyleni⁃mine,PEI)吸附siRNA/miRNA 分子[11],分析其对巨噬细胞的作用,为高效诱导巨噬细胞向M2 极化提供新的思路。
1 材料和方法
1.1 主要材料 十六烷基三甲基溴化铵(cetyltri⁃methylammonium bromide,CTAB)、氢氧化钠(sodi⁃um hydroxide,NaOH)、正硅酸四乙酯(tetraethyl or⁃thosilicate,TEOS)、PEI(Sigma⁃Aldrich,美 国);RAW264.7 小鼠巨噬细胞系(ATCC,美国);DMEM培养基、胎牛血清、青⁃链霉素、PBS 缓冲液、胰蛋白酶等细胞培养试剂(Hyclone,美国);MTT、DAPI、CUR、DiI(MP Biomedical,美国);总RNA 提取试剂盒、反转录试剂盒、引物以及实时定量PCR 试剂盒(TaKaRa,日本);抗体(ABCAM,英国);靶向GAP⁃DH 的siRNA(siGAPDH,正义:5'⁃CACUCAAGAUU⁃GUCAGCAATT⁃3';反义:5'⁃UUGCUGACAAUCUUG⁃AGUGAG⁃3')、siRNA 阴性对照(siNC,正义:5'⁃UU⁃CUCCGAACGUGUCACGUTT⁃3';反义:5'⁃ACGUG⁃ACACGUUCGGAGAATT⁃3')、miRNA 错 配 序 列 阴性 对 照(miNC,5'⁃CGGUACGAUCGCGGCGGGAU⁃AUC⁃3')、miR⁃130a⁃3p 反义寡核苷酸(ASO,5'⁃AUGCCCUUUAACAUUGCACUG⁃3')等购于上海吉玛公司。
1.2 MSN 的合成及共递送系统构建
依据文献报道[12],采用常规sol⁃gel 方法制备MSN,即称取0.50 g CTAB 溶解于250 mL 去离子水中,加入1.75 mL 2.0 mol/L NaOH 溶液,充分搅拌并升温至80 ℃,反应30 min,然后在剧烈搅拌下加入2.75 mL TEOS,至产生白色沉淀后加入2.50 mL 乙酸乙酯,80 ℃反应2 h。室温陈化过夜后,离心,用超纯水和无水乙醇各洗涤3 次,抽滤,真空干燥,得到白色固体。将白色固体转入三口烧瓶中,加入50 mL 无水乙醇,2.5 mL 盐酸充分分散后,80 ℃加热回流24 h 去除CTAB,用超纯水和无水乙醇各洗涤3 次,抽滤。真空干燥,得到MSN。
1.3 CUR 与siRNA 的共递送系统构建
采用MSN 构建CUR 与质粒的共递送系统进行本实验[13]。取上述MSN 粉末4 mg 加入到1 mL 含有1 mg CUR 的无水乙醇溶液中,使药物与载体的质量比分别为1∶4,超声至MSN 完全分散后,磁力搅拌过夜,将溶液于15 000 rpm/min 离心15 min,沉淀经无水乙醇洗涤3 次,真空干燥,得到淡黄色颗粒CUR@MSN。称取1 mg PEI 加入到1 mL 无水乙醇中超声分散,将CUR@MSN 加到PEI 溶液中,水浴超声30 min,离心,无水乙醇洗涤3 次,得到(CUR@MSN)PEI。将(CUR@MSN)PEI 颗粒加入到siRNA(2 μmol/L)溶液中,水浴超声30 min,离心,无水乙醇洗涤3 次,得到(CUR@MSN)PEI/siRNA 纳米共递送系统。将样品分散在无水乙醇中,滴在铜网上,在室温下干燥数小时后行透射电镜观察。
将适当稀释后的CUR@MSN 吸取1 mL 转入透析袋内(截留相对分子质量为3 500),密封后放入200 mL 含0.2% SDS 的PBS 溶液中,于37 ℃恒温水浴;分别于2、4、6、12、24、48、72 h 后取透析液1 mL,同时补充等量的释放介质;通过在酶标仪A425nm处测定吸光度的方法测定CUR 含量,并计算累积释放百分比。
1.4 细胞培养及活力检测
采用巨噬细胞株RAW264.7 细胞进行后期细胞实验,细胞培养液为DMEM+10%胎牛血清+1%青⁃链霉素,在37 ℃、体积分数5% CO2培养箱中培养,在细胞密度为80%左右时进行胰蛋白酶消化传代。将细胞接种在96 孔板内,将(CUR@MSN)PEI 纳米粒用培养基倍比稀释后处理细胞24 h,随后换成普通培养基再培养24 h 后进行MTT 法检测细胞活力。具体方法:将5 mg/mL 的MTT 溶液与培养液预混合(MTT:培养液=1∶4)后加入细胞培养孔内,每孔200 μL,在细胞培养箱孵育4 h 后取出,酶标仪A580nm测吸光度值,并用未处理细胞进行数据标准化。
1.5 细胞转染效果观察
将RAW264.7 细胞接种在玻底培养皿内,使用FAM 荧光标记的siRNA 进行细胞转染实验,在转染结束后与含DiI 染料的培养基孵育15 min,吸弃培养液后使用4%多聚甲醛固定细胞10 min,PBS清洗两遍,DAPI 染色液染色5 min,PBS 清洗两遍后,使用激光共聚焦扫描显微镜观察细胞转染效果。
另一方面,在转染结束后立刻采用胰蛋白酶消化,离心收集细胞团块,采用2.5%戊二醛与4%多聚甲醛混合液(1∶1)进行固定后采用1%锇酸进行二次固定,酒精梯度脱水后环氧树脂包埋、钻石刀超薄切片,铜网捞片后行醋酸铀/柠檬酸铅染色,透射电镜观察纳米粒在细胞内的分布情况。
1.6 实时定量PCR(RT⁃qPCR)检测GAPDH 的沉默效率
将RAW264.7 细胞接种在6 孔板内,按照转染复合物的不同分成4 组,即实验组:CUR⁃siGAPDH共递送体系[(CUR@MSN)PEI/siGAPDH];对照组:未处理细胞(未进行转染)、空白载体[(CUR@MSN)PEI]、CUR⁃siNC 共递送体系[(CUR@MSN)PEI/siNC]。将各组转染复合物加入细胞培养液中,十字混匀;转染4 h 后更换为普通培养基继续培养;在48 h 后,采用RNA 提取试剂盒提取细胞内总RNA,采用PrimerScript RT reagent Kit 试剂盒逆转录合成cDNA,再以cDNA 为模板使用SYBR Premix Ex Taq 试剂盒进行RT⁃qPCR 反应,以β⁃actin 作为内参基因,检测GAPDH 的沉默效率。其中,小鼠GAPDH 引物序列为:上游引物5'⁃GTTGTCTCCTGC⁃GACTTCA⁃3',下游引物5'⁃GGTGGTCCAGGGTTTC⁃TTA⁃3’';小鼠β⁃actin 引物序列为:上游引物5'⁃CT⁃GTCCCTGTATGCCTCTG⁃3',下游引物5'⁃ATGTCA⁃CGCACGATTTCC⁃3'。
1.7 CUR⁃ASO 共递送体系对巨噬细胞M2 型极化的影响
将RAW264.7 细胞接种在6 孔板内,按照转染复合物的不同分成4 组,即实验组:CUR⁃ASO 共递送体系[(CUR@MSN)PEI/ASO],其中ASO 为miR⁃130a⁃3p 反义寡核苷酸;对照组:未处理组(未进行转染)、空白载体[(CUR@MSN)PEI]、CUR⁃miNC 共递送体系[(CUR@MSN)PEI/miNC]。转染4 小时后更换为普通培养基继续培养,在3 d 后采用West⁃ern Blot 进行Arg⁃1 蛋白的表达检测。主要步骤为采用RIPA 加蛋白酶抑制剂裂解细胞,提取总蛋白,煮沸10 min 后与上样缓冲液混合进行SDS⁃PAGE 凝胶电泳,转膜后与Arg⁃1、β⁃actin 的一抗孵育过夜,次日与HRP 偶连的二抗孵育后发光液成像。采用ImageJ 分析条带灰度值。
1.8 统计学分析
用Graphpad Prism 8 软件处理数据,计量数据采用均数± 标准差表示,不同组间比较采用单因素方差分析,两两比较采用SNK⁃q 检验,校验水准为α=0.05。
2 结 果
2.1 纳米粒表征观察
采用透射电镜观察可见制备的MSN内部呈现出经典的孔道结构,整体纳米粒直径80~100 nm(图1a);在吸附CUR 小分子后,MSN 内部的孔道结构被低电子密度物填充,提示CUR 成功吸附进入MSN 内部(图1b);在CUR@MSN 表面吸附PEI 大分子后,纳米粒的直径有所增大,内部孔道结构仍被低电子密度物充填,表明CUR@MSN 表面形成了PEI 涂层,且MSN 内部的CUR 未发生泄漏(图1c)。通过紫外分光光度计检测,可计算出MSN 对CUR 的包封率和载药率分别为65.74%、16.57%,加载siRNA 的效率约94%。通过累积释放曲线可见,CUR 被MSN 吸附进入内部后呈现出缓慢释放的特征,在最初24 h 内可累积释放约20%,随后的释放量较小(图1d),表明MSN 可实现CUR 的长期携带作用。
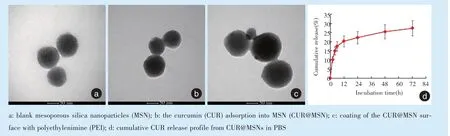
Figure 1 Transmission electron microscopy observation during the preparation of nanoparticles and release curve after the adsorption of CUR图1 纳米粒制备过程的透射电镜观察及吸附CUR 后的释放曲线
2.2 细胞毒性筛查
分别采用不同浓度的(CUR@MSN)PEI 纳米粒处理细胞,采用MTT 法检测细胞活力以评估对细胞安全的浓度范围。可见在高浓度的纳米粒处理下,细胞活力受到显著抑制,当粒子浓度低于10 μg/mL 时细胞活力在90%以上,未表现出明显的细胞活力抑制(图2),提示该浓度为安全范围,在后期细胞功能实验中均采用了10 μg/mL 进行转染。
2.3 细胞转染观察
共聚焦显微镜观察可见绿色荧光聚集在红色区域内、蓝色细胞核周围(图3a),表明纳米粒可被细胞摄取;同时,透射电镜可直接观察到细胞内的(CUR@MSN)PEI/siRNA 纳米颗粒聚集(图3b),放大观察后可见纳米粒已部分发生降解(图3c)。以上结果表明纳米共递送系统可成功转染进入巨噬细胞,且转染效率较高。

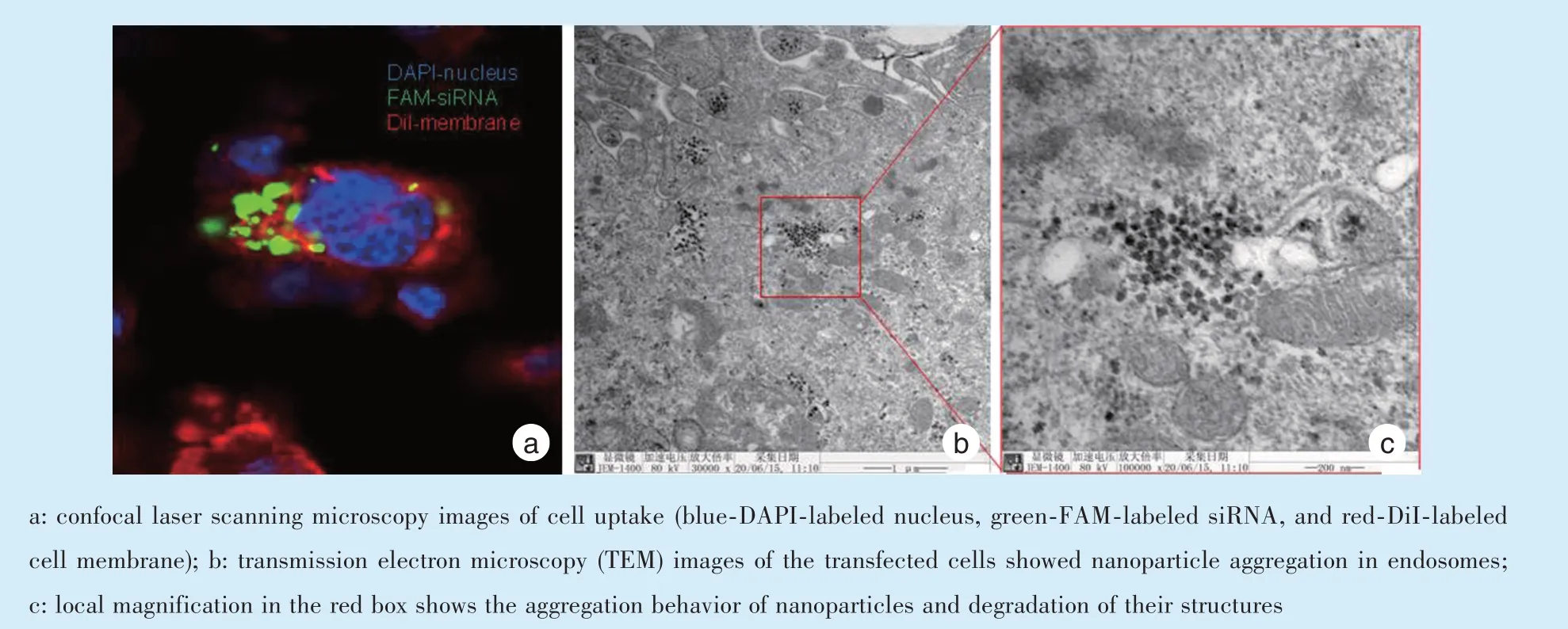
Figure 3 (CUR@MSN)PEI/siRNA nanoparticle uptake observed by confocal microscopy and TEM图3 共聚焦显微镜和透射电镜观察细胞摄取(CUR@MSN)PEI/siRNA 纳米粒
2.4 靶基因沉默效率检测
采用RT⁃qPCR 检测GAPDH 的表达变化,在导入GAPDH 的siRNA 后细胞表达GAPDH 下降到未转染细胞的0.16 倍,即沉默效率高达80%以上(图4,P <0.01);阴性对照siRNA 则未表现出显著的沉默效果,表明沉默作用是来自于siRNA 的序列匹配作用。这表明(CUR@MSN)PEI 载体系统可安全高效的递送siRNA/miRNA,进而达到沉默靶基因的目的。
2.5 巨噬细胞M2 极化检测
与未处理的细胞组相比,(CUR@MSN)PEI 和(CUR@MSN)PEI/miNC 组的Arg⁃1 表达提升2~3 倍(P <0.05),(CUR@MSN)PEI/ASO 组的Arg⁃1 表达提升约8 倍(P <0.05)(图5)。
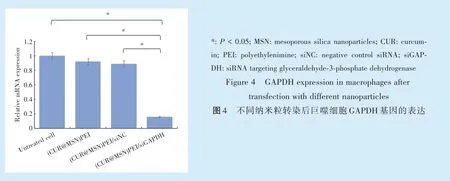

Figure 5 Arg⁃1 protein expression in macrophages after the co⁃delivery of CUR⁃ASO图5 CUR⁃ASO 共递送系统转染巨噬细胞后Arg⁃1 蛋白的表达
3 讨 论
3.1 基于MSN 的共递送系统制备与表征
种植体在植入体内后巨噬细胞首先发生反应调控无菌性炎症反应,其不同的极化方向对于炎症转归至关重要,其中M2 方向极化的巨噬细胞可分泌白介素⁃4(interleukin⁃4,IL⁃4)、血小板衍生生长因子(platelet derived growth factor,PDGF)等细胞因子,促进局部组织的愈合过程,有利于骨结合的快速建立。因此通过调控种植体周围巨噬细胞向M2 方向极化促进其与机体的整合效果已经成为了新的策略。CUR 是较为经典的诱导M2 极化的小分子药物,为更好发挥CUR 作用,本研究制备出基于MSN 的CUR 与siRNA 的共递送系统,通过透射电镜观察证实了合成的MSN 呈现经典的规整孔道结构,内部和表面分别成功吸附了小分子CUR 和高分子PEI。由于MSN 的特殊结构,其内部孔道可用于吸附分子量较小的分子,表面可吸附分子量较大的分子,是共递送载体的理想选择[11]。
多种纳米载体系统均可用于共递送系统的构建,除了常用的MSN,还包括有机阳离子聚合物等[14],这种共递送系统能够克服单一药物的不足,发挥两种药物优势的协同作用。例如针对肿瘤耐药问题,其机理为耐药肿瘤细胞内高表达P⁃glyco⁃protein(Pgp)分子,有研究采用介孔硅内部负载抗肿瘤药物多柔比星,表面携带针对Pgp 的siRNA,在进入细胞后可沉默细胞Pgp 分子的表达,从而解除细胞对多柔比星的耐药性[15]。
3.2 纳米粒的毒性及细胞摄取
在成功构建出纳米递送系统后,纳米材料的细胞毒性是进行生物应用首先考虑的问题。与文献报道类似[16],本研究制备的负载CUR 和PEI 修饰后的MSN 表现出剂量依赖性的细胞毒性反应,在合适的浓度范围内纳米粒对细胞活力的影响可以被控制在理想范围,实现细胞内的药物递送作用。在安全的浓度范围内处理细胞,通过激光共聚焦显微镜可观察到细胞摄取了siRNA 进入胞浆,进一步采用透射电镜可直接在胞浆内观察到纳米粒聚集存在;此外,在透射电镜观察中可见纳米粒聚集性存在于囊泡结构中,表明细胞是通过内吞作用进行纳米粒摄取,这与前期报道的MSN 进入细胞的途径一致[17];部分纳米粒的结构变得模糊,表明在被细胞摄取后逐渐发生了降解,MSN 的这种快速降解特性也为其生物安全性提供了保证[18]。
3.3 靶基因沉默效率及诱导巨噬细胞M2 极化
采用阳性对照GAPDH siRNA 进行细胞转染是评估转染载体常用的方法,可避免靶基因的差异性对载体的转染效率产生影响,能够更好地确定转染载体的作用效率[19]。本实验构建的(CUR@MSN)PEI 纳米载体可高效实现RAW264.7的GAPDH 沉默,为负载功能性的核酸奠定了基础。
在诱导实验中,单纯导入CUR 即可诱导巨噬细胞M2 极化标志物Arg⁃1 的高表达,证实了CUR可通过激活PPARγ 受体诱导巨噬细胞的M2 极化。Arg⁃1 是最为常用的巨噬细胞M2 极化的标志分子,本课题组前期研究也以该分子的变化作为衡量巨噬细胞M2 极化程度的关键指标,是较为简便、可靠的方法[20]。Su 等[9]采用高通量组学的方法系统性探索了巨噬细胞极化过程中miRNA 的调控网络,发现miRNA⁃130a⁃3p 在巨噬细胞M2 极化中表达下调,进一步分析发现其靶基因恰为PPARγ受体,即当巨噬细胞向M2 方向极化时miR⁃NA⁃130a⁃3p 发挥内源性抑制PPARγ受体的作用,当引入miRNA⁃130a⁃3p 的反义序列ASO 后,可解除其抑制效应,进一步提升巨噬细胞M2 向极化。本研究中也发现,相比单纯CUR 的处理组,同时转染ASO 后可显著进一步诱导巨噬细胞M2 极化标志物Arg⁃1 的表达,表明ASO 与CUR 可能发生了协同作用,共同提升了巨噬细胞向M2 极化。
综上所述,本研究成功构建了基于MSN 的CUR 与siRNA/miRNA 的共递送系统,在安全浓度范围内可高效转染巨噬细胞,发挥小分子药物与核酸的协同作用,高效诱导巨噬细胞M2 向极化。这为诱导种植体周围巨噬细胞的M2 向极化,促进骨结合的建立提供了新策略。
【Author contributions】Li PH wrote the article. Lang K analyzed the data. Song W designed the study. All authors read and approved the final manuscript as submitted.
猜你喜欢
现代医药卫生(2022年20期)2022-11-20
发明与创新(2022年31期)2022-11-03
中国药学药品知识仓库(2022年10期)2022-05-29
航天电子对抗(2022年2期)2022-05-24
中国医药科学(2022年5期)2022-05-05
北京大学学报(医学版)(2022年1期)2022-04-14
北京航空航天大学学报(2021年9期)2021-11-02
雷达学报(2021年1期)2021-03-04
中国药学药品知识仓库(2021年18期)2021-02-28
船海工程(2020年2期)2020-06-08
