
心房颤动与线粒体功能障碍
2021-03-10 09:17尚帅张玲汤宝鹏
中国心脏起搏与心电生理杂志 2021年1期
尚帅 张玲 汤宝鹏
心房颤动(简称房颤)是临床上最常见的持续性心律失常之一,但房颤的诱发与维持的机制尚不清楚。线粒体作为心房肌细胞的能量工厂,既是心房肌细胞内氧化磷酸化和合成三磷酸腺苷(ATP)的主要场所,又参与心肌细胞离子如Ca2+、Na+和K+的稳态调节,对心脏收缩、能量代谢和电活动有重要作用。房颤时发生线粒体DNA 及生物合成异常、能量代谢障碍及酶的改变、线粒体反应活性氧类增多及氧化应激增强、Ca2+等多种离子通道异常、线粒体形态结构改变等[1]。Wiers ma等[2]对房颤患者的心房活检显示线粒体功能障碍,表现为ATP水平异常、线粒体应激伴侣上调和线粒体网断裂,同时冠状动脉旁路移植术(CABG)的患者中,心房肌细胞线粒体功能障碍者有更高的术后房颤发生率[3]。这些研究提示房颤可能与线粒体的结构和功能障碍密切相关。笔者从以下几个方面论述二者之间的联系。
1 房颤与线粒体的生物合成异常
在线粒体的生物合成过程中,涉及到核基因组和mt DNA 的转录调控途径,通常我们用mt DNA 含量来反映线粒体的生物合成功能。其生物合成过程中的关键调节因子叫做过氧化物酶体增殖物激活受体γ辅助活化因子1ɑ(PGC-1ɑ)。有研究发现,在快速起搏引起的成年兔房颤模型中存在线粒体发生的PGC-1α/NRF-1/Tfam 途径的转录下调有助于心房代谢重塑,最终扰乱了线粒体功能,在体外循环后的手术后房颤组心房组织PGC-1ɑ显著降低[4]。而非诺贝特通过调节PPAR-α/sirtuin 1/PGC-1α途径来抑制房颤的房室代谢重塑[5]。同时也有研究表明,PGC-1β缺陷小鼠心脏线粒体功能障碍会继发年龄依赖性房性心律失常[6]。在研究长时间心跳停止的猪模型中血管内低温对线粒体生物发生的影响实验中,血管内低温通过放大线粒体生物发生因子PGC-1α等的表达来提高线粒体活性和生物发生,缓解了心肌和线粒体功能障碍[7]。除此之外,同源结构域转录因子2(Pitx2)通过其基因调控网络促进线粒体功能,在Pitx2缺乏的情况下,由于电和结构重构,动作电位持续时间缩短、传导缓慢和触发活动发生,左右心房之间的电异质性增加,从而促进折返。在小鼠心肌细胞中成对的Pitx2 丢失的情况下更容易发生房颤,而氟卡胺通过阻止自发性钙释放和增加波长,对Pitx2 受损所致的房颤具有抗心律失常的作用[8]。
2 房颤与线粒体DNA
线粒体具有自身的遗传物质,即双链环状线粒体DNA,其自身可以独立进行复制、转录和翻译部分线粒体蛋白质。Hiona等[9]通过小鼠模型研究发现,mt DNA 突变可导致线粒体功能障碍,生物合成下降,ATP产生减少。另有研究发现,在慢性房颤患者的mt DNA 控制区和编码区均发生mt DNA 突变,包括小缺失和组织特异性长度异质性突变,以及外周血白细胞线粒体mt DNA4977缺失突变明显增加,并且与心房的结构重构和电重构有关[10]。Tsuboi等[11]研究发现,mt DNA 缺失与人房颤时腺嘌呤核苷酸的减少有关,mt DNA 缺失7.4kb的患者房颤患病率明显高于无缺失的患者。同时mt DNA 缺失组右心房ATP、ADP、AMP和腺嘌呤核苷酸总量均低于无缺失组。Montaigne等[3]在对手术后房颤患者的研究中发现,基因表达谱分析确定了发生房颤的患者术前心房组织中线粒体/oxphos基因簇的普遍下调,为患者线粒体与房颤等心律不齐之间的联系提供了新的见解。除此之外,Soltész等[12]研究中从60例房颤患者和72例健康对照者中抽取外周血,从血液和血浆中分离DNA;从无细胞血浆中分离出外泌体,然后提取外泌体封装的DNA。两个研究组的mt DNA 拷贝数在外周血,无细胞血浆和外泌体甚至不同性别和年龄之间均无显著差异。结果表明,房颤患者的mt DNA 拷贝数与健康者没有差异。但Zhang等[13]检测了485例窦性心律CABG 患者外周血mt DNA 拷贝数。手术后房颤患者mt DNA 平均拷贝数显著高于窦性心律患者。分析表明,线粒体DNA 拷贝数预测手术后房颤具有良好的敏感性和特异性,是手术后房颤的独立危险因素。
3 房颤与线粒体的能量代谢障碍及酶的改变
在心肌细胞中,90%以上的ATP 是在线粒体内膜氧化磷酸化生成的,用于满足自身的机械活动,维持细胞膜和肌浆网上各种离子通道、转运体和酶的功能,以实现心肌细胞的正常电活动。在心肌能量代谢严重下降时,各种依赖ATP的离子通道或转运蛋白的功能及酶的活性将受损[14]。Anderson等[15]对244例心脏外科手术患者的右心耳组织研究发现单胺氧化酶(MAO)和还原型辅酶Ⅱ(NADPH)氧化酶产生的活性氧速率比完整的偶联线粒体高10倍。其结果表明,MAO 是决定人心房肌氧化还原平衡的重要因素,该酶与手术后的房颤风险增加有关。有研究发现线粒体是心房肌细胞对能量需求反应的关键,房颤时线粒体功能障碍和重塑导致ATP耗竭,其中线粒体中电子转运链的整体功能活性降低了30%、氧化磷酸化复合物(琥珀酸脱氢酶和细胞色素C氧化酶)受损、活性氧增加。在心脏外科手术中,一些围手术期因素可以影响线粒体功能,包括由于暴露于体外循环和手术组织创伤而增加的全身炎症反应。线粒体功能障碍最直接影响术后心肌收缩力,也易导致房颤等心律失常。而钠-葡萄糖共转运蛋白2抑制剂依帕列净具有恢复线粒体功能、改善电和结构重塑以及预防房颤的能力[16-19]。
4 房颤与线粒体反应活性氧类产生及氧化应激
反应活性氧类(reactive oxygen species,ROS)是一类未被完全还原的含氧分子,由O2得到单电子产生的超氧阴离子(O2-),以及再逐步接受电子而生成的过氧化氢H2O2、羟自由基(·OH)等组成。房颤可引起线粒体内呼吸链受损、电子泄漏增加,导致线粒体内ROS 产生增多,增多的ROS可以促进线粒体通透性转变孔(mPTP)和内膜阴离子通道(I MAC)的开放,使线粒体网络中的ΔΨm 同时去极化,进一步促进了ROS的产生。而过量的ROS对细胞脂质和蛋白质产生有害反应,导致心肌细胞损伤功能障碍,并且可以通过相关的信号分子间接地调节离子通道的转运蛋白功能,例如ROS 敏感激酶,包括钙调蛋白依赖性蛋白激酶(Ca MKⅡ)、c Src和蛋白激酶C(PKC),或者通过氧化还原敏感的转录因子,如NFκB来损害心肌兴奋性。同时线粒体功能受损会使NADH、ADP、乳酸盐、H+增多及谷胱甘肽(GSH)耗竭,共同加重心房肌细胞氧化应激,进一步促进房颤的发生与维持,而抑制I MAC 或应用心肌还原型GSH、NADPH 氧化酶(NOx)抑制剂可以预防房颤等心律失常[17,20-21]。
有研究发现,房颤患者表现出较高的氧化应激,增加了线粒体ROS 的产生,同时心房中的2 型ryanodine 受体(Ry R2)被氧化,而线粒体氧化应激后产生的ROS会使肌/内质网Ca2+-ATPase(SERCA)活性被抑制、肌膜ATP敏感性 钾 通 道(sarcole mmal ATP-sensitive potassiu m ,sarc KATP)被激活及Ry R2开放增加,进而促进肌质网中Ca2+通过Na+-Ca2+交换体(NCX)的释放,导致肌质网中Ca2+储存耗尽、胞质中Ca2+超载,引起心房肌细胞作用电位延长、后去极化延迟、收缩功能障碍,且具有细胞内Ca2+泄漏的小鼠表现出心房Ry R2氧化增加,线粒体功能障碍,ROS产生增多和房颤敏感性增加。表明Ry R2和线粒体ROS生成的变化在房颤的发展中形成了恶性循环,减少线粒体ROS的产生从而减弱心房舒张期肌质网Ca2+的泄漏可以预防房颤[22-23]。
5 房颤与线粒体相关Ca 2+等多种离子通道平衡紊乱
心肌细胞胞质中Ca2+浓度的变化是心肌细胞收缩的关键,而心肌细胞线粒体内储存的大量Ca2+作为细胞内的Ca2+库,对其维持正常的生理功能有着重要的作用。Ca2+主要通过线粒体Ca2+单向转运体(MCU)进入基质,而线粒体内Ca2+外流主要由线粒体Na+-Ca2+交换体(mNCX)、线粒体通透性转变孔(mPTP)及Ca2+反向转运体所介导[24]。有研究提示,心脏组织中微区结构的丧失或局部Ca2+失调通常与氧化应激、收缩功能障碍和房颤等心律失常有关,在房颤的电学重构中产生的过多线粒体Ca2+,可以激活mPTP导致线粒体ROS产生增多诱导凋亡性细胞死亡;可以激活ATP酶,加速ATP的消耗;激活核酶来引起染色体的损伤[25]。除此之外,增多的Ca2+可以抑制K+-H+交换体、触发KCa和mito KATP通道,增加K+流入和线粒体基质的体积导致线粒体肿胀。同时在线粒体功能障碍时,会降低INa峰值、下调Cx43使折返型心律失常的传导异常和倾向增加;增加晚INa、降低复极化电压门控K+通道(Kv)电流使复极受损,导致APD 和EAD 延长、心肌电异质性增加及心律失常敏感性增加[1]。房颤时线粒体及心肌离子通道的变化见图1。Brown和O′Rour ke[26]及Zhou等[27]研究表明线粒体的能量状态对心肌细胞膜动作电位的影响在很大程度上由sarc KATP介导,且sarc KATP离子流强弱与线粒体内膜电位(ΔΨm)的波动有关。线粒体功能障碍时,ΔΨm 降低,ATP生成减少,导致sarc KATP 通道开放,心肌局部电活动传导减慢,不均一性增加,易于形成折返诱发心律失常。有研究表明,在发生手术后房颤的44%患者中,对钙诱导的mPTP开放的敏感性增加与手术后房颤相关,而线粒体功能障碍可通过部分阻断或下调MCU 来阻止线粒体Ca2+内流的增加而减弱房颤重构[2-3]。同时,心房纤维化在房颤的病理生理中也有一定作用,而利伐沙班能够通过抑制Ca2+进入而降低了心房纤维母细胞的迁移能力,从而调节心房成纤维细胞的活性和房颤的发生[28]。也有学者通过计算机模拟表明,肌浆网钙泄漏导致减少的外向电流IK1和增加的内向钠钙交换电流之间的不平衡导致自发去极化,并且通过抑制肌浆网钙泄漏和逆转重构的IK1,可以潜在地抑制这些致心律失常的触发因素[29]。
6 房颤与线粒体的形态结构改变
房颤时除了发生心房电重构和神经体液重构外,心房肌细胞线粒体形态结构也发生变化。张玲等[30]在成年比格犬用快速右心房起搏(600次/分)建立48 h及8周房颤模型,取心房肌组织行透射电镜检查,观察到48h房颤组心肌细胞肌纤维间质增宽,间质部线粒体聚集;8周房颤组肌原纤维排列紊乱,肌节长短不一,线粒体肿胀变长,体积增大,基质变淡,嵴异形明显。提示房颤引起心房肌线粒体发生多种结构改变。但目前仍缺乏相关对其变化机制进行探讨的具体研究。
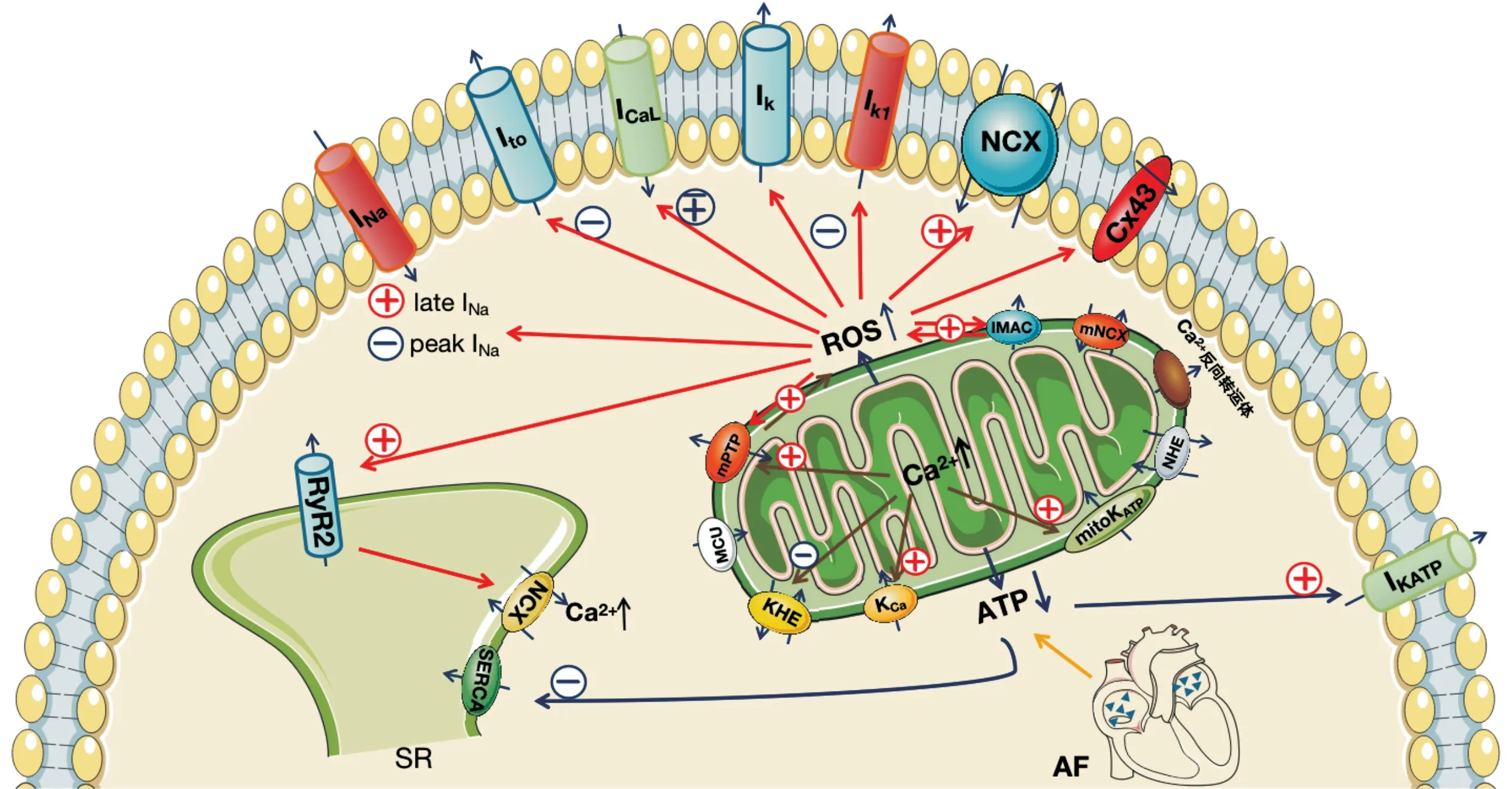
图1 房颤时线粒体及心肌离子通道的变化
7 总结
线粒体是心房肌细胞能量代谢的关键一环,并且在氧化应激、维持细胞离子稳态及细胞内信号传导等方面有着重要的作用。房颤时会引起心房肌线粒体能量代谢障碍、离子稳态失衡、mt DNA 及其形态功能改变等,给我们预防及治疗房颤提供了新的启示,保护及改善线粒体功能,或可成为预防及治疗房颤的潜在靶点。
房颤时会引起线粒体功能障碍,导致ROS 产生增加、ATP合成减少和线粒体Ca2+增多。ROS升高可抑制INa峰值、Ito、IK、IK1活性、促进线粒体通透性转变孔(mPTP)和内膜阴离子通道(I MAC)的开放及Cx43 的下调、增强晚INa,Na+-Ca2+交换体(NCX)和Ry R2的活性。同时Ry R2开放增加能够促进肌质网中Ca2+通过NCX 的释放,导致肌质网(SR)中Ca2+储存耗尽、胞质中Ca2+超载。线粒体功能障碍继发的ATP 产量减少可抑制SERCA 的活性并增加肌膜KATP通道的活性。Ca2+主要通过线粒体Ca2+单向转运体(MCU)进入基质,而线粒体内Ca2+外流主要由线粒体Na+-Ca2+交换体(mNCX)、线粒体通透性转变孔(mPTP)及Ca2+反向转运体所介导。房颤的电学重构中产生的过多线粒体Ca2+,可以激活mPTP 导致线粒体ROS 产生增多,抑制K+-H+交换体(KHE)、触发KCa和mito KATP 通道,增加K+流入。
猜你喜欢
中西医结合心脑血管病杂志(2022年4期)2022-03-11
中西医结合心脑血管病杂志(2022年2期)2022-02-15
少男少女·小作家(2021年5期)2021-08-04
体育科学(2018年12期)2019-01-04
科学之谜(2018年9期)2018-12-17
戏剧之家(2018年35期)2018-02-22
中国医药导报(2017年34期)2018-01-29
青年文学家(2017年20期)2017-07-29
试题与研究·高考理综生物(2016年4期)2017-03-28
作文评点报·高中版(2017年9期)2017-03-27
