
DEPDC5基因突变相关的癫痫综合征的诊治分析
2021-03-09 00:21龙绮婷张玮张翠荣尚丽柴英冯亚梅龚德山刘兴洲
癫痫与神经电生理学杂志 2021年1期
龙绮婷,张玮,张翠荣,尚丽,柴英,冯亚梅,龚德山,刘兴洲
DEPDC5基因突变可导致多种癫痫综合征,大部分的患者发作类型为局灶性发作,其中一部分患者通过结构及功能影像学检查可发现局灶性异常,例如广泛大脑皮层发育畸形和局灶性皮层发育不良[1]等。其相关的癫痫综合征包括:家族性多变局灶性癫痫(familial focal epilepsy with variable foci,FFEVF)[2-3]、家族性颞叶内侧癫痫(familial mesial temporal lobe epilepsies,FMTLE)[3]、常染色体显性遗传夜间额叶癫痫(autosomal dominant nocturnal frontal lobe epilepsy,ADNFLE)[4]、常染色体显性遗传癫痫并听觉障碍(autosomal dominant epilepsy with auditory features,ADEAF)[5]、婴儿痉挛(infantile spasms)[6]和良性癫痫伴中央颞区棘波(benign epilepsy with centrotemporal spikes,BECTS)[7]等。临床表现为多数在婴儿或儿童期起病,局灶性发作类型多见,夜间发作常见,大部分患者为药物难治性癫痫,一部分患者存在癫痫家族史;神经系统查体发现大部分患者的精神运动发育正常,部分患者可存在轻度认知功能下降或孤独症样表现[2,8];结构功能成像可发现广泛大脑皮层发育畸形、局灶性皮层发育不良、巨脑回畸形和多微脑回畸形[9-10]等。本文通过总结3个DEPDC5基因突变家系的临床表现、头颅结构及功能影像学和基因学等特点,阐述DEPDC5基因突变导致的癫痫为常染色体显性遗传,不完全外显,其癫痫发作表现多样,即便在同一家系中也有可能出现不同类型的癫痫发作,在临床工作中需要结合临床表现、癫痫家族史、结构功能成像和基因学筛查等,对于部分局灶性癫痫患者通过外科手术可以达到满意效果。现报道如下。
1 资料和方法
1.1 临床资料
本文总结2017年至2019年就诊于上海德济医院的3个DEPDC5基因突变家系的临床资料,包括病史、查体、脑电图、核磁共振、头颅18氟-脱氧葡萄糖标记正电子发射计算机断层成像(PET-CT)和基因学等结果。
A家系先证者为女性,30岁,右利手,生长发育正常,本科毕业,工商管理专业。14岁首次发作,发作表现为先兆(恐惧,心慌)→ 心率增快 → 喉部发声 → 右手摸索,左上肢僵硬不动,每次发作持续20 s到1 min左右,睡眠期发作占70%,发作频率10~12次/月。病程中只有1次继发全身强直阵挛发作。起病后曾服用托吡酯、卡马西平、丙戊酸镁、氯硝西泮等治疗均无效,目前服用奥卡西平600 mg/早、晚,左乙拉西坦750 mg/早、晚治疗。既往史无特殊。家族史:患者父亲有癫痫病史,12岁起病,30岁至今无发作,具体表现不详;患者姐姐的女儿有失神发作,患者姑姑的外孙有强直发作(图1A)。神经系统查体无特殊。韦氏全量表94分,韦氏记忆75分。
B家系先证者为男性,19岁,右利手,生长发育轻度落后同龄儿童,语言少,有孤独症样表现,初二辍学。3岁首次发作,发作表现为先兆(头部不适,难以描述)→ 眨眼、右侧凝视→ 四肢僵硬,每次持续10~20 s,清醒期发作占100%,每日发作5~7次。起病后曾服用拉莫三嗪、卡马西平、氯硝西泮、丙戊酸钠、唑尼沙胺等治疗均无效,目前服用拉莫三嗪100 mg/早、中、晚,唑尼沙胺150 mg/早、中,300 mg/晚。17岁在外院行“左额中下回+右额中回+胼胝体前2/3切除术”,术后发作表现及频率同术前。家族史:患者的舅舅有癫痫病史,36岁起病,表现为全身强直阵挛发作(图1B)。神经系统查体:孤独症样表现,眼神交流及语言表达少,其余无特殊。韦氏全量表47分,韦氏记忆<50分。
C家系先证者为女性,5岁零8个月,右利手,生长发育同其他同龄儿童,幼儿园中班在读。11月龄时首次发作,11月龄至2岁零5个月龄发作表现为双眼上翻、四肢僵硬,数秒缓解,成串发作,每日1~2串,每串10余次。2岁零5个月龄后发作表现:眨眼、颜面潮红、四肢僵硬,随后抖动数下,每日1~2次,睡眠中发作多见。起病后曾服用丙戊酸钠、左乙拉西坦、拉莫三嗪、托吡酯等治疗均无效,曾给予生酮饮食治疗无效,目前服用丙戊酸钠9 mL/早、晚,托吡酯50 mg/早、晚,拉莫三嗪12.5 mg/早治疗。既往史、家族史无特殊(图1C)。神经系统查体无特殊。韦氏智力及记忆不能配合。

图1 A家系中箭头所示为先证者,发作表现为先兆(恐惧,心慌)→植物神经症状→复杂运动→左上肢肌张力障碍,先证者的父亲有癫痫病史,12岁起病,30岁至今无发作,具体表现不详;先证者姐姐的女儿有失神发作;先证者姑姑的外孙子有强直发作。B家系中箭头所示为先证者,发作表现为先兆(头部不适)→眨眼→右侧凝视→左上肢强直,先证者的舅舅有全身强直阵挛发作,先证者的母亲携带基因。C家系中箭头所示为先证者,发作表现为①痉挛发作;②双侧非对称强直发作,先证者的父亲携带基因。
1.2 研究方法
1.2.1 头皮脑电图
对先证者进行头皮脑电图检查记录间歇期放电及发作期症状学表现。监测时间至少24 h,包括清醒期及睡眠期,至少记录3次以上惯常发作。
1.2.2 头颅核磁共振
完善头颅核磁共振检查,序列包括Flair成像(3 mm层厚,0间距),T1WI成像(1 mm层厚,0间距),了解有无脑结构异常。
1.2.3 头颅PET-CT
完善头颅PET-CT(1 mm层厚,0间距),了解有无脑功能代谢异常。
1.2.4 高通量全外显子测序与Sanger测序验证
在获得先证者及家属的知情同意后,取其4 mL静脉血进行全外显子测序。对先证者家属取静脉血进行Sanger测序验证。
1.2.5 立体定向颅内电极脑电图
根据头皮脑电图及结构、功能成像检查,3个家系的先证者考虑为局灶性癫痫,因需确定致痫区范围,征求患者家属同意后行立体定向颅内电极植入,并监测间歇期及发作期放电情况确定致痫区范围。
1.2.6 手术切除及病理
3个家系的先证者,根据立体定向颅内电极脑电图,结合结构及功能成像决定致痫区范围,并行致痫区切除术,术后标本送病理检查。
2 结果
2.1 头皮脑电图
A家系先证者头皮脑电图间歇期放电提示右侧半球弥漫性棘慢波,以右侧前头部为著(图2),发作期症状学表现为先兆(恐惧,心慌)→植物神经症状(心率增快60~105次/min)→复杂运动(右上肢重复摸被子、整理衣服、挪髋等)→左上肢肌张力障碍,脑电图提示右侧半球棘慢波。B家系先证者头皮脑电图间歇期提示右侧额-中央区棘慢波、多棘慢波(图3),发作期症状学表现为先兆(头部不适,难以描述)→眨眼→右侧凝视→左上肢强直,脑电图提示右侧额-中央区棘慢波→棘慢波。C家系先证者头皮脑电图间歇期提示右侧半球弥漫性棘慢波、多棘慢波,以右额区为著(图4),发作期症状学表现为①11月龄时记录到痉挛发作,脑电图提示全部性;②2岁零5个月龄后表现为双侧非对称强直发作(左侧肢体为著)→眼球震颤→强直阵挛(累及眼睑、面肌、四肢),脑电图提示右额区棘慢波→弥漫性棘慢波和多棘慢波。
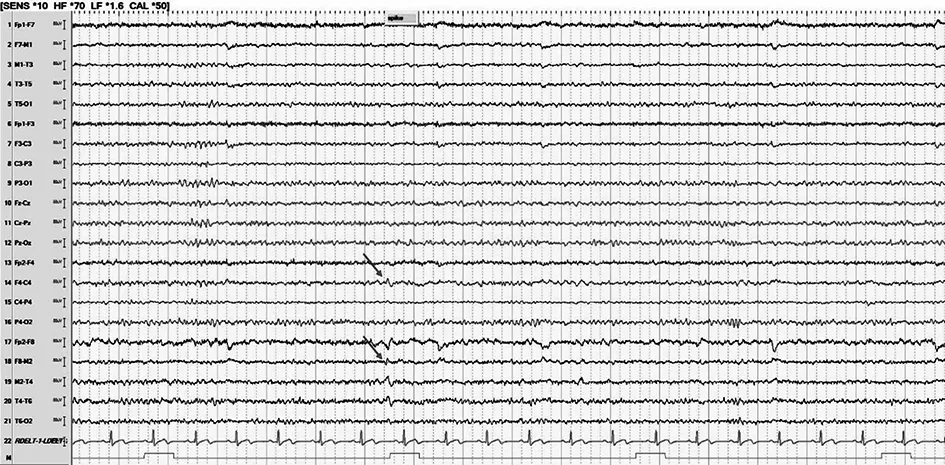
图2 A家系先证者的脑电图。清醒期背景可,双侧后头部节律10~11 Hz,调节调幅可,箭头示右侧半球弥漫性棘慢波,以右侧前头部为著。

图3 B家系先证者的脑电图。清醒期可见双侧前头部缺损节律,双侧后头部节律9~10 Hz,调节调幅可,箭头示右侧额-中央区多棘慢波。

图4 C家系先证者的脑电图。清醒期背景可,双侧后头部节律9~10 Hz,调节调幅可,箭头示右侧半球弥漫性棘慢波、多棘慢波,以右额区为著。
2.2 头颅核磁共振及PET-CT
A家系先证者头颅核磁共振未发现明显脑结构异常,PET-CT提示右侧眶额区、前扣带回、右侧额叶内侧面、双侧颞极、颞叶内侧结构和岛叶低代谢(图5)。B家系先证者头颅核磁共振提示右侧额上回、额上沟发育异常,PET-CT提示右侧额上回、额叶内侧面及额上沟低代谢(图6)。C家系先证者头颅核磁共振提示右侧额上回、额上回旁中间沟(superior frontal para-midline sulcus,SFPS)发育畸形,PET-CT提示右侧额上回、扣带回低代谢(图7)。
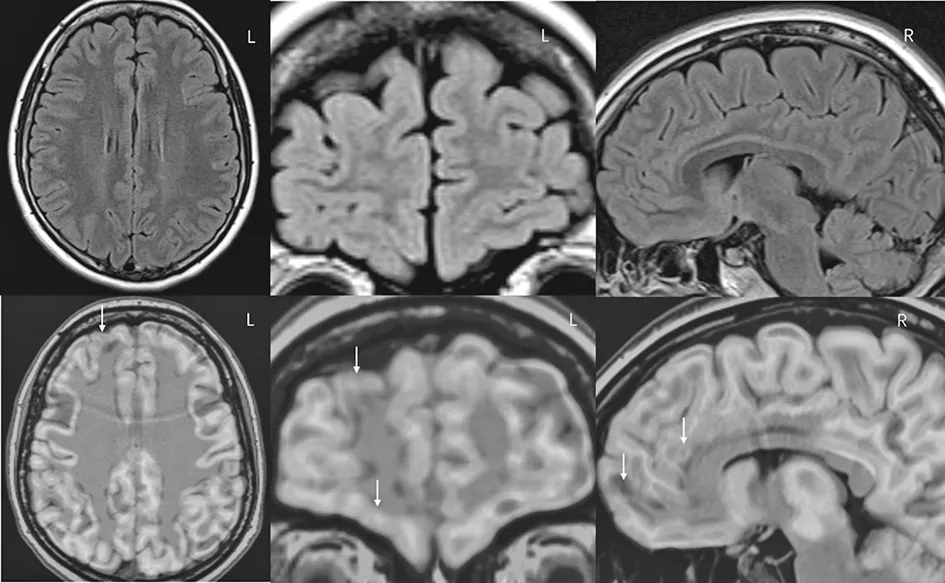
图5 A家系先证者的头颅核磁共振未见明显脑结构异常,头颅PET-CT与核磁共振融合后(箭头所示)提示右侧眶额区、右侧额叶内侧面,双侧颞极、颞叶内侧结构低代谢。
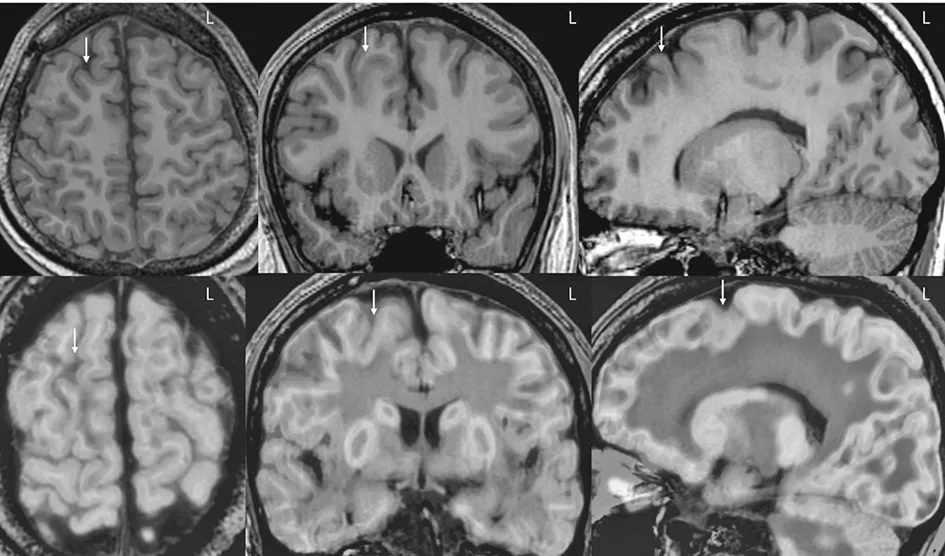
图6 B家系先证者在外院的术前头颅核磁共振(箭头所示)提示右侧额上回、额上沟发育异常,头颅PET-CT与核磁共振融合(箭头所示)提示右侧额上回、额叶内侧面及额上沟低代谢。
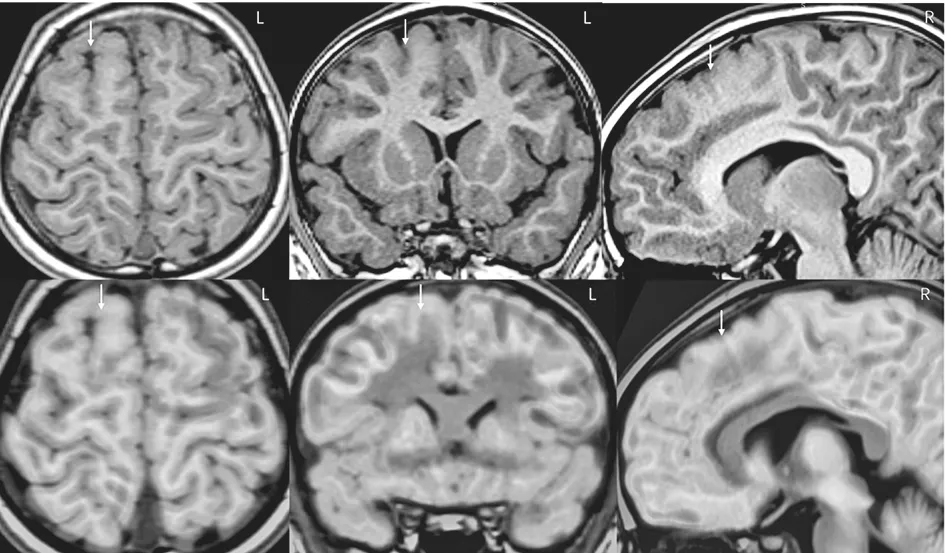
图7 C家系先证者的头颅核磁共振(箭头所示)提示右侧额上回、旁中间沟发育畸形,头颅PET-CT与核磁共振融合(箭头所示)提示右侧额上回、扣带回低代谢。
2.3 高通量全外显子测序与Sanger测序验证
A家系先证者22号染色体(NM_001242 896.1)区域存在缺失突变,cDNA水平c.489_491delGTT,使得所编码的蛋白质第164 位Phe发生缺失p.(Phe164del),功能学研究表明该变异引起DEPDC5通路信号减弱,抑制了GATOR-1复合物形成,从而增强TORC1激酶活性。先证者的父亲在相同位点发现突变。有文献报道在多个家系的家族型多变性局灶性癫痫患者中检测到该变异,考虑该变异为病理性变异[2]。
B家系先证者22号染色体(NM_001242896.2)区域存在错义突变,cDNA水平c.4620C>A,该外显子位于基因最后一个外显子,为罕见变异,但有报道认为该变异之前的无义突变 Gln1536为致病性变异[2],因此考虑该变异为病理性变异。先证者的母亲在相同位点有变异,但无临床表现,考虑为不完全外显。先证者的舅舅有癫痫发作病史,但拒绝验证基因位点,推测其舅舅可能携带相同基因。
C家系先证者22号染色体位置chr22:32211008存在缺失突变c.1476delA,导致外显子exon21所编码的蛋白序列p.Arg492Argfs*25发生移码突变,在其后的25位氨基酸位置发生截断突变。患者的父亲在相同位点有突变,但无临床表现。该位点无既往的文献报道,结合患者临床表现及基因学特点,考虑为病理性变异。
2.4 立体定向颅内电极脑电图
根据头皮脑电图,结构、功能影像学检查,3个家系的先证者考虑为局灶性癫痫,行立体定向颅内电极植入,并监测间歇期及发作期放电。
A家系先证者考虑致痫区位于右侧额叶,需要鉴别右侧颞叶,行立体定向颅内电极植入,植入方案如下(图8①②)。术后监测颅内电极放电情况,清醒期眶额区触点B′6-8持续棘慢波,伴有高频放电;杏仁核、颞极、海马触点有少量放电。睡眠期前扣带回触点B′1-2持续多棘慢波,伴有高频放电;眶额区触点Y′1-2近持续多棘慢波;杏仁核及海马触点少量放电。发作前期眶额区触点B′6-8及Y′1-6棘慢波及高频放电。脑电图发作起始从眶额区触点B′6-8,Y′1-6起始,早期扩散至眶额区触点X′1-2。考虑致痫区位于右侧眶额区、前扣带回、额叶内侧面(图8)。

图8 ①②为A家系先证者的右侧半球电极植入方案;③为核磁共振轴位,显示B′1-2触点位置位于前扣带回;④为核磁共振轴位,显示B′电极入点位于后眶额区,靶点位于前扣带回; ⑤为核磁共振矢状位,显示Y′电极入点位于额中回,靶点位于后眶额区;⑥为核磁共振冠状位,显示X′1-2触点位置位于内眶额区。
B家系先证者考虑致痫区位于右额上沟、额上回,需要明确致痫区的范围(后界及外侧界),行立体定向颅内电极植入,植入方案如下(图9①②)。术后监测颅内电极放电情况,清醒期额上沟触点Y′5-10持续棘慢波,伴有高频放电;前辅助运动区、中央前沟触点有同步性棘慢波放电。发作前期额上沟触点Y′5-8及U′5-8棘慢波、多棘慢波,伴有高频放电。脑电图发作起始从Y′5-8、U′5-8起始,早期扩散至前辅助运动区触点Z′1-6、S′6-7、U′1-4、Q′1-3。考虑致痫区位于右侧额上沟、额上回(图9)。

图9 ①②为B家系先证者的右侧半球电极植入方案;③④为核磁共振轴位,显示Y′6-8触点位于额上沟内侧壁,Z′1-4触点位于前辅助运动区;⑤为核磁共振轴位,显示S′电极入点位于中央前回,穿中央前沟到达靶点辅助运动区;⑥为核磁共振冠状位,显示U′电极入点位于额中回,穿额上沟到达靶点前辅助运动区。
C家系先证者考虑致痫区位于右额上回、右额上回旁中间沟,需要明确致痫区的范围(后界,内侧面的下界),行立体定向颅内电极植入,植入方案如下(图10①②)。术后监测颅内电极放电情况,清醒期额叶旁中间沟触点C′5-7、G′4-6持续性棘慢波,伴有高频放电;前扣带回触点G′1-4、Q′1-2持续性棘慢波。发作前期额叶旁中间沟触点C′5-7、G′4-6及前扣带回触点G′1-4棘慢波、多棘慢波,伴有高频放电。脑电图发作起始弥漫,受累电极触点包括额叶旁中间沟、前扣带回、额上回、额下回。考虑致痫区位于右额上回旁中间沟、额上回(图10)。

图10 ①②为C家系先证者的右侧半球电极植入方案;③为核磁共振轴位,显示C′6触点位于额上回旁中间沟;④为核磁共振轴位,显示G′6触点位于额上回旁中间沟的沟底,C′1触点位于中扣带回;⑤为核磁共振轴位,显示Q′4触点位于额上回,G′2触点位于前扣带回;⑥为核磁共振矢状位,显示Q′1触点位于前扣带回。
2.5 手术及病理
根据立体定向颅内电极脑电图监测结果,结合结构及功能成像,3位家系先证者均行致痫区切除术。A家系先证者切除范围包括右侧直回、眶额区(内眶额区、后眶额区)、额叶内侧面及额上回、前扣带回。B家系先证者切除范围包括右侧额上回、额上沟,外侧面完整切除额上沟,内侧面后界到旁中央沟前壁。C家系先证者切除范围包括右侧额上回、额上回旁中间沟、前扣带回、中扣带回,内侧面后界到旁中央沟前壁。切除方案见图11。3位家系先证者手术过程顺利,术后无不适,无后遗症表现。A家系先证者的病理结果提示胶质细胞增多,B家系及C家系先证者的病理结果提示局灶性皮层发育不良(focal cortical dysplasia,FCD)IIA型。3位家系先证者术后随访至今均无发作。A家系先证者术后至今2年余无发作,已逐渐减药,调整药物为奥卡西平600 mg/早、晚,左乙拉西坦500 mg/早、晚,减药期间无发作。B家系先证者术后至今1年余无发作,继续按照术前药物服药,目前所`服药为拉莫三嗪100 mg/早、中、晚,唑尼沙胺150 mg/早、中,300 mg/晚。C家系先证者术后至今2年余无发作,已逐渐减停托吡酯,目前服药为丙戊酸钠9 mL/早、晚,拉莫三嗪12.5mg/早,减药期间无发作。

图11 ①②分别为A家系先证者在核磁共振冠状位及三维成像的切除方案,范围包括右侧直回、眶额区(内眶额区、后眶额区)、额叶内侧面及额上回、前扣带回;③④分别为B家系先证者在核磁共振轴位及三维成像的切除方案,范围包括右侧额上回、额上沟,外侧面完整切除额上沟,内侧面后界到旁中央沟前壁;⑤⑥分别为C家系先证者在核磁共振轴位及三维成像的切除方案,范围包括右侧额上回、额上回旁中间沟、前扣带回、中扣带回,内侧面后界到旁中央沟前壁。
3 讨论
DEPDC5基因位于22号染色体长臂22q12.2-q12.3,其编码的1 604氨基酸与NPRL2及NPRL3共同组成GATOR1 (GTPase-activating protein activity towards rags) 复合物,抑制mTORC1(mechanistic target of the rapamycin complex 1)通路。mTORC1具有丝氨酸-苏氨酸激酶活性,对下游网络蛋白进行磷酸化,从而调节重要的细胞功能,如蛋白合成、细胞生长及迁徙和分化等。DEPDC5基因突变导致GATOR1复合物减少,mTORC1活性增强,mTOR通路激活,可能会导致神经细胞之间的突触联系方式发生改变,神经元兴奋性增高,但具体如何导致脑结构改变机制目前尚不明确。已有报道表明, NPRL2或NPRL3突变导致的癫痫综合征表现与DEPDC5相关的癫痫综合征类似[8,11],提示GATOR1复合物为重要的影响因素。动物实验表明,在DEPDC5基因纯合突变的老鼠,mTORC1的活性增强,可导致严重的颅脑畸形等[12]。在本文的3位家系先证者中,A及B家系的先证者致病位点已有报道与家族性多变局灶性癫痫相关,C家系的先证者基因发生移码突变,导致后续的氨基酸发生截断突变,考虑为病理性变异。
DEPDC5基因突变相关的癫痫综合征为常染色体显性遗传,外显率为60%~70%[2],一般具有家族性发病特点,部分个体为新发突变[6],具体比例不详。其相关的癫痫综合征包括:家族性多变局灶性癫痫综合征(FFEVF)、家族性颞叶内侧癫痫(FMTLE)、常染色体显性遗传夜间额叶癫痫(ADNFLE)、常染色体显性遗传癫痫并听觉障碍(ADEAF)、婴儿痉挛和良性癫痫伴中央颞区棘波(BECTS)等。在本研究中,高通量全外显子测序对3个家系中共9例进行全外显子检测,其中携带DEPDC5基因突变者6例,有癫痫发作者4例,外显率为67%,与文献报道一致。
FFEVF首次在DEPDC5基因相关的癫痫综合征中被报道,目前已发现超过80个DEPDC5基因位点突变可导致该综合征,其主要特征为患者的首次发作年龄多在婴幼儿期,癫痫发作症状学由致痫区部位决定,发作类型为局灶性发作,其中以额叶及颞叶最为多见。具有癫痫家族史。在同一家系中,不同的亲属其致痫区部位可以不一致,发作类型不一。因此,FFEVF的诊断基于家族层面,不能单从个体来诊断。在本研究中A家系具有明显的家族聚集特点,其中4例亲属有癫痫病史,4例亲属的癫痫发作表现不一,考虑为家族性多变局灶性癫痫综合征。在本研究中B家系中除了先证者外,先证者的舅舅也有癫痫病史,两者的癫痫发作类型不同,结合病史及结构功能成像,考虑为家族性多变局灶性癫痫综合征。
DEPDC5基因突变相关的癫痫综合征患者,以往文献报道大部分智力发育正常,一部分可出现轻度智力落后、心理或行为异常、孤独症样表现。在本文中,A家系及C家系先证者精神运动发育基本正常。而B家系先证者,其智力发育稍差于同龄儿童,韦氏全量表分值低于同龄人,且合并孤独症样表现。
既往的文献报道,发现DEPDC5基因突变相关的癫痫综合征常见的大脑皮层发育畸形为FCD Ⅱ型、巨脑回畸形、双侧对称性外侧裂综合征等[13]。在本研究的B、C家系先证者,通过结构核磁共振成像均发现右侧额叶广泛皮层发育畸形。在本研究的A家系先证者,通过功能成像PET-CT发现右侧额、颞、岛叶广泛低代谢,提示致痫区范围较大。3位家系先证者通过颅内电极脑电图监测明确致痫区范围后均行手术切除,其中B、C家系先证者病理结果提示FCD ⅡA型,与文献报道一致。
本文通过总结3个家系的临床及辅助检查资料,揭示DEPDC5基因突变相关的癫痫综合征多数具有癫痫家族史,大部分的患者精神运动发育正常,其癫痫发作类型以局灶性发作多见,单药治疗效果普遍欠佳,多数患者为药物难治性癫痫。通过结构及功能成像往往可以发现存在大脑皮层发育畸形。值得一提的是,其致痫区往往范围较大,与FCD不同,可以累及脑区或者脑叶。本研究的3位家系先证者通过立体定向颅内电极脑电图,结合结构及功能成像决定致痫区范围,发现致痫区并不仅仅限于脑沟或脑回。完整地切除致痫区后,3个家系的先证者术后随访至今均无发作,提示DEPDC5基因突变的癫痫综合征通过手术干预可以达到良好的效果。但对于癫痫发作类型为全部性癫痫,或者双侧半球致痫、多脑区致痫的患者,可能难以通过手术达到无发作。在临床工作中,诊断需要结合癫痫家族史、发作的症状学、视频脑电图记录间歇期及发作期脑电图、结构及功能成像以及基因学检查等,以明确为哪种类型的DEPDC5基因突变癫痫综合征,选择合适的治疗方法。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22
影像研究与医学应用(2020年17期)2020-08-20
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
读者(2016年18期)2016-08-23
广东海洋大学学报(2015年4期)2016-01-13
听力学及言语疾病杂志(2015年5期)2015-12-24
首都医科大学学报(2015年4期)2015-12-16
重庆医学(2015年12期)2015-03-05
郑州大学学报(医学版)(2015年2期)2015-02-27
