
二维铁电体CuCrP2S6的第一性原理研究
2021-03-08 08:21翟元玮冯敏王玉芳
光散射学报 2021年2期
翟元玮,冯敏,王玉芳
(南开大学物理科学学院,天津 300071)
1 引言
自从人类偶然中发现了铁电性质之后,铁电体在之后的百年间经历了繁荣的发展,无数的科研人员将目光聚焦在了这种集众多优良性质,如铁电性、非线性光学、压电性等等于一身的多功能材料。铁电系统作为一种新兴的应用材料,在许多方面有着光明的应用前景,具有广泛的工业和商业用途,其涉及的领域从高介电常数电容器到压电换能器、热释电传感器等的发展。以往对于铁电材料的研究焦点都集中在三维材料上,如BiFeO3等钙钛矿结构。然而,随着纳米技术日趋成熟,磁电器件的尺寸缩小到只有几个纳米。随着尺寸逐渐缩小,材料的比表面积逐渐增大,这意味着在表面的原子所占比例越来越高,这会带来一系列负面的影响——如表面原子的表面能会不断升高且表面张力也越来越大。如此一来,位于表面的原子很不稳定,有许多悬空键,导致电子器件的性能和稳定性较差,且同时量子隧穿效应越来越显著,器件的漏电流增大[1]。由此一来,可以完美解决上述问题的二维材料进入人们的视野。这种对范德华材料的迷恋引起了人们对层状金属硫(硒)磷酸盐(MTSPs)家族的兴趣。MTSPs最早发现于20世纪80年代末,采用A1+B3+P2X6化学计量比。一层带有磷二聚体的金属阳离子位于两层硫或硒(S,Se)原子之间。许多MTSPs在较低温度下表现出反铁电(AFE)或铁电(FE)有序和/或铁磁(FM)或反铁磁(AFM)有序。这种同时具有FE和FM有序性或多铁性的材料在自旋电子学和非易失性存储器等领域有着丰富的研究、应用价值。[2]
2 结构优化
本文重点关注CuCrP2S6晶体的二维结构与性质,利用materials studio中的切割晶胞工具,我们切出其中一层来观察该晶体的结构。并在垂直ab面的方向设置15 Å的真空层,用来屏蔽层间的范德华作用。
利用VASP软件,为了获得合理的结构,几何结构和离子被充分弛豫,收敛标准为任何原子上的受力都小于0.01 ev/Å而总能量的收敛条件设置为相邻两次迭代的体系总能量之差小于1×10-4eV。综合考虑到计算精度与所耗时间的平衡,设置平面波基组的截止能量为400 eV,指定将用于采样布里渊区的布洛赫矢量(K点)为11×7×1的Monkhorst-Pack网格。
本文采用基于GGA的Perdew-Burke-Ernzerh(PBE)交换关联泛函[3]计算原子与电子结构。为了获得合理的结构,本文采用了分步结构优化的方法,第一步选择的优化参数为ISIF=7,只优化晶格基矢。第二步用ISIF=2,在保持晶格基矢的条件下优化原子的坐标,反复几次直到基矢和原子位置都达到稳定值即达到收敛标准。晶体结构如图1所示

图1 CuCrP2S6晶体的基本结构
在CuCrP2S6单层膜结构中,6个S原子形成周期环,Cu、Cr原子和P原子对交替排布在环的中心。在该晶体中,Cu有两种不同的位置:Cu原子位于单层结构的同一侧导致正负电荷中心不重合,进而产生垂直平面的电偶极子(极化),对应晶体的铁电相;Cu原子交替位于单层结构的两侧,产生两个方向相反的电偶极子,但总的电偶极子为0,对应晶体的反铁电相。铜离子在铁电相和反铁电相之间的位移距离约为2.73Å。
在这里,只有通过2个铜原子的相对位置关系,才能体现出铁电相和反铁电相的不同,即自发极化的原因,而菱形原胞则不能表现出这一点。所以下文中所比较的能量值,都是基于矩形晶胞的选取。
铁电相和反铁电相的俯视图以及侧视图分别由图2和图3所示。

图2 单层反铁电相CuCrP2S6(a)俯视图(b)侧视图

图3 单层铁电相CuCrP2S6(a)俯视图(b)侧视图
我们对体相和单层的铁电相和反铁电相均进行了结构优化,发现两者的晶格常数几乎没有区别。本文将优化后的晶格常数与文献实验结果进行了对比,如表1所示。由于本文模拟的是二维结构,所以对比时忽略了c轴。
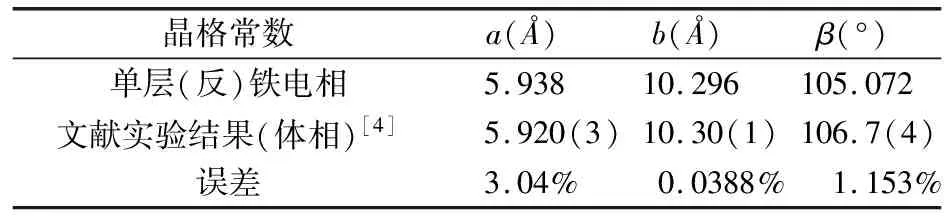
表1 结构优化与实验数据对比
3 静态能量计算
在这一步中,我们固定原子的位置并且不再进行原子弛豫,调整系统中的电子以达到系统能量最低。通过静态自洽计算,我们可以获得体系达到收敛条件后的总能量。
考虑到了磁性的问题,所以本文在输入文件中为每个原子都给定了初始的磁矩,在计算过程中VASP会自动优化到合适的磁矩大小。本文根据核外电子排布情况和在晶体中的价态,设定了硫原子初始磁矩为6μB,硫原子初始磁矩为3μB,铜原子初始磁矩为0μB,磷原子初始磁矩为1μB。
由于还有此晶体中含有过渡元素Cu和Cr,密度泛函DFT理论(LDA或GGA)给出的结果会低估d或f电子的强库仑斥力,包括自旋电子对之间的相互作用以及自旋电子与内层电子的斥力。所以本文考虑了对DFT-GGA理论的一种修正,称为Hubbard U或者GGA+U修正。[5]此模型的基本假设是,强关联的d或f电子(可在紧束缚单粒子基础上描述)受现场准组相互作用的影响。其中最重要的是Hubbard参数Ueff。本文测试了Ueff=0,1,2,3下的自洽计算的结果,来比较他们的能量大小及能量差。

表2 在不同Ueff下,铁电相与反铁电相的能量对比
之后,本文采用了CI-NEB方法模拟计算了铁电相与反铁电相之间相变的能量变化曲线以及能垒[6]。CI-NEB是一种在已知初态和末态结构之间寻找鞍点和最小能量路径的方法。此方法会不断优化相变路径上的中间结构,找到每个结构最低的能量,并且在此过程中保证与相邻结构的相等间隔。
本文计算了反铁电相AFE和铁电相FE之间的相变过程,如图4和图5所示。

图4 由反铁电相相变为铁电相过程的能量随铜原子位置变化曲线

图5 由铁电相相变为反铁电相过程的能量随铜原子位置变化曲线
我们可以发现,两相之间相变的能量变化路径曲线对于两个相变方向来说是相同的。如果铁电相相变为反铁电相,需要跨过一个76meV的能垒。这说明铁电相虽然能量较高,但足够大的能垒使得它可以稳定存在,这确保了它广阔的应用前景。
4 性质计算
在进行静态自洽计算后,我们就得到了体系的电荷密度分布,我们可以利用其计算我们需要的性质。
4.1 能带
能带计算中取的高对称点如图6所示。

图6 能带计算所选取的布里渊区高对称K点示意图
其中,Γ(0,0,0),X(0.5,0,0),Y(0,0.5,0),S(0.5,0.5,0),X'(-0.5,0,0),Y'(0,-0.5,0),S'(-0.5,-0.5,0)。在Γ-Y(Y')、Y(Y')-S(S')、S(S')-X(X')、X(X')-Γ这八段特殊点路径中间,每段取40个K点来计算能带结构。为了测试GGA+U对能带结果的影响,依然测试了Ueff=0,1,2,3下的结果。在这里只列出Ueff=3下的计算结果,因为其能带结构和带隙最接近文献中类似体系的结果[4]。

图7 单层CuCrP2S6的能带结构(a)反铁电相-自旋向上(b)反铁电相-自旋向下(c)铁电相-自旋向上(d)铁电相-自旋向下
根据对比,我们可以发现,Ueff对自旋向下的能带的能隙影响较大,对自旋向上的能隙以及总能隙影响很小。通过对比文献,当Ueff=3时,能隙最接近,所以我决定以此为正式结果。
对于铁电相和反铁电相,计算出的能隙分别为1.1797 eV和0.9851 eV,且均为间接带隙。因此,单分子膜CuCrP2S6是半导体,可能是理想的光催化剂。
4.2 态密度(DOS)和分波态密度(PDOS)
为了进一步探究CuCrP2S6晶体的电子结构,本文进一步计算了其总态密度和分波态密度。设置态密度展宽为0.05 eV,同时加入Hubbard U修正Ueff=3。

图9 铁电相自旋向上情况下,费米能级附近能带、分波态密度和总态密度对比图

图10 反铁电相自旋向上情况下,费米能级附近能带、分波态密度和总态密度对比图
本文重点关注了价带顶和导带底的贡献情况,如图8-图11所示:对于低能部分的导带而言,无论是铁电相还是反铁电相,也无论自旋向上或者自旋向下,主要贡献来自Cr的d电子态和S的p电子态;而对于价带顶来说,自旋向上时主要贡献为 Cr、Cu的d电子态和S的p电子态,而且铁电相中Cu的贡献明显减少。自旋向下时主要贡献为Cu的d电子态和S的p电子态。

图11 铁电相自旋向下情况下,费米能级附近能带、分波态密度和总态密度对比图
4.3 声子谱
接下来,为了验证结构的稳定性,本文计算了单层结构的声子谱。首先,我将晶体扩胞为2×2×1的超胞,设置平面波基组的截止能量为400 eV,总能的收敛条件为相邻两次迭代的体系总能量差小于1×10-8eV。声子谱结果如图15所示。

图12 单层CuCrP2S6结构反铁电相的声子谱
对于单层反铁电相来说,最低点的虚频在5个波数,gamma点的虚频在2个波数,单位为cm-1。由于二维晶体结构普遍存在一定的虚频,所以这个结果可以一定程度上说明单层CuCrP2S6晶体结构是足够稳定的,后续可以考虑继续扩胞,消除这些虚频。
由于选取的晶胞中含有20个原子,所以共有60条声子谱曲线(振动模式),其中有3支声学支格波,57支光学支格波。gamma点处最低的三条曲线位于声学频率范围内,即是声学支格波,其余为光学支格波。
单层CuCrP2S6二维反铁电相晶体空间群对称群为P21,点群为C2。根据晶格振动模对称性因子群分析方法,其布里渊区中心点晶格振动模的对称性为:Γsym=20A+30B;扣除三个声学模,57个光学模的对称性为:Γopt=29A+28B。由第一性原理计算得到布里渊区中心点振动模的对称性以及频率值列表3中。

表3 单层CuCrP2S6二维晶体反铁电相布里渊区中心点振动模的频率(cm-1)和对称性
4.4 光学性质
我们可以通过计算材料的频率相关的复介电函数ε(ω),得到该半导体的一系列非常有用的线性光学性质。
ε(ω)=ε1(ω)+iε2(ω)
(1)
其中,ε1(ω)和ε2(ω)分别是复介电函数的实部与虚部,ω则表示光子的频率。
由于二维结构真空层厚度的任意性,所以本文利用光学电导率σ2D(ω)来表征二维薄板的光学性能。基于麦克斯韦方程,三维光学电导率可表示为
σ3D(ω)=i[1-ε(ω)]ε0ω
(2)
式中,ε(ω)是与频率相关的复介电函数,ε0是真空的介电常数,ω是入射波的频率。平面内二维光导率与相应的σ3D(ω)分量直接相关
σ2D(ω)=cσ3D(ω)
(3)
其中,c为模拟二维体系的层厚度,显然此式与真空层厚度无关。
假设正入射时,对独立二维晶体的归一化反射率R(ω)、透射率T(ω)和吸光度A(ω)与光的偏振无关,那么
(4)
(5)
(6)

由此,我们就可以画出二维结构的介电函数、吸收率、透射率以及反射率。在这里,只放出反铁电相的相关曲线图,因为与铁电相的几乎无差别,唯一区别下文会提到。
由于介电函数是二阶张量,所以吸收率、透射率以及反射率也均为二阶张量。图例中即为张量下标。
通过图我们可以发现,在光子能量为5~10 eV区间,单层CuCrP2S6有一个吸收峰,这说明该单层晶体对125~250 nm的紫外光的吸收最强,此时透射率最低,另外在整个光子能量计算范围内吸收率都非常低。

图13 单层反铁电相CuCrP2S6介电函数实部

图14 单层反铁电相CuCrP2S6介电函数虚部

图15 单层反铁电相CuCrP2S6光吸收率

图16 单层反铁电相CuCrP2S6光反射率

图17 单层反铁电相CuCrP2S6光透射率
并且,通过对比铁电相和反铁电相的上述光学性质,可以发现对于反铁电相来说,在光子能量为8.6 eV时,吸收率曲线有一个较高的峰,此峰在铁电相中并不存在,吸收率低了5%左右。
4.5 自发极化
根据现代极化理论[8],极化的相对值才是有实际意义的。所以计算极化值时需要一个参考相。由于不好构造对称结构,本文选择利用极化向下(-z轴)的结构作为参考相,计算极化向上(+z轴)结构的自发极化强度。即
pz=(p↑z-p↓z)/2
(7)
其中,P↑z和P↓z分别表示极化向上和向下结构的z方向自发极化强度。
另外,二维结构的极化强度通常定义为单位面积内的总电偶极矩。因此,在这里现代理论中的极化量子变为
(8)
由于极化绝对值的多值性,我们还需要把离子极化计算结果的数据人为地加上极化量子的整数倍,才可以将计算得到的结果重新投影到[-1,1)ΔPi的区间内,这样计算出的相对极化值才有意义且准确。
自发极化的计算结果如表3所示。

表4 自发极化计算结果
其中,piz表示 Z轴方向的离子偶极矩,pez表示 Z轴方向的电子偶极矩遵循计算公式
pz=(p↑z-p↓z)/2
(9)
pz=[(p↑iz+p↑ez)-(p↓iz+p↓ez)]/2
(10)
可以得到,对于极化向上的结构,在z轴方向总偶极矩为
pz=0.0967e.A.
(11)
而该二维结构的自发极化强度为

=1.2588pC/m
(12)
其中,a和b为晶体的晶格常数。
5 结论
通过第一性原理理论模拟计算,证明了二维CuCrP2S6单层结构是一种理想的光催化剂半导体,其铁电自发极化性质可以稳定存在。本文计算了其能带结构并探究了费米能级附近的电子态贡献,同时计算了其光学性质以及自发极化强度。该二维铁电体可在实现二维铁电光伏和铁电存储方面有广阔的应用前景。
猜你喜欢
分子催化(2022年1期)2022-11-02
无机材料学报(2022年6期)2022-08-25
小天使·聪聪画刊(2021年2期)2021-09-10
科学(2020年4期)2020-11-26
汽车零部件(2020年10期)2020-11-09
汉语世界(The World of Chinese)(2019年6期)2019-09-10
电子制作(2019年15期)2019-08-27
测控技术(2018年9期)2018-11-25
苏州科技大学学报(自然科学版)(2017年1期)2017-03-20
上海大中型电机(2017年4期)2017-02-06
