
长链非编码RNA LINC00628通过竞争性结合miR-145促进肺腺癌细胞吉非替尼抵抗
2021-03-08 01:22刘晨阳张成杨婕孙韬
临床肺科杂志 2021年3期
刘晨阳 张成 杨婕 孙韬
表皮生长因子受体酪氨酸激酶抑制剂(epidermal growth factor receptor tyrosine kinase inhibitor,EGFR-TKI)作为EGFR阳性肺癌患者治疗的一线靶向用药,能显著延长晚期患者生存时间,改善患者生活质量。然而EGFR-TKI抵抗的形成,严重影响了其疗效[1],其形成的机制复杂,涉及EGFR突变、c-Myc基因扩增及肿瘤干细胞等。非编码RNA作为表观遗传的重要执行者,与恶性肿瘤的发生发展密切相关。成熟的miRNA通过与其互补的mRNA结合,进而促进靶mRNA的降解或抑制其翻译[2]。miR-145可靶向c-Myc、Oct4及AKT等原癌基因,在多种恶性肿瘤中发挥抑癌作用[3-5],因此,揭示miR-145的调控机制对于肿瘤的早期诊断及治疗方案的评估至关重要,长链非编码RNA(long non-coding RNA,lncRNA)是长度>200nt的非编码RNA,其可作为内源竞争RNA(competing endogenous RNAs,ceRNA),抑制miRNA的生物学效应,参与调控肿瘤细胞的恶性行为及药物抵抗[6-7]。本研究通过生物信息技术发现,与miR-145潜在结合的lncRNA LINC00628,并对其与miR-145的结合作用及与肺腺癌细胞EGFR-TKI抵抗的关系进行了探究。
资料与方法
一、实验材料
1 实验细胞及试剂 人肺腺癌细胞系PC9(上海生科院细胞资源中心),RPMI-1640培养基及胎牛血清(FBS,美国Gibco),吉非替尼(Gefitinib,美国MCE),LINC00628 小干扰RNA(small interference RNA LINC00628,siLINC)及随机序列对照表达载体(上海吉凯基因),LINC00628过表达载体、miR-145过表达载体及空白载体(上海汉恒生物),合成miR-145 NC及mimics(上海汉恒生物科技),转染试剂Lipofectamine 2000(美国Thermo Fisher),psiCHECK2载体及Luciferase Assay Reagent II试剂盒(美国Promega),CCK-8试剂(日本同仁化学),RNAiso Plus、PrimeScript RT reagent Kit及SYBR Green I(日本Takara),GAPDH、c-Myc及AKT抗体(美国Proteintech)。
2 临床组织来源 收集2019年7月至2019年12月我院普胸外科54例肺腺癌患者肿瘤组织及癌旁组织,取材后取部分组织置于液氮罐中保存,54例患者中男性30例,女性24例;年龄36~71岁,中位年龄51岁;包括高分化腺癌11例,中低分化腺癌43例;Ⅰ~Ⅱ期患者25例,Ⅲ~Ⅳ期患者29例,所有患者均为首次发现的原发性肺腺癌,术前未经抗肿瘤治疗。
二、实验方法
1 细胞培养及分组 PC9细胞采用90% RPMI-1640+10% FBS培养于37℃+5% CO2+100%相对湿度的环境中。将PC9细胞传代后分为7组:对照组(Control组)、吉非替尼抵抗组(Gefitinib resistance,GR组),干扰组(small interference LINC00628,siLINC组)、干扰对照组(siControl组),空白组(Blank组)、过表达组(miR-145组)及回复组(Rescue组)。GR组采用0.1、0.2、0.4、0.8、1.6、3.2 μmol/L吉非替尼逐步诱导PC9细胞GR的形成,每个浓度梯度维持培养2周以上,直至细胞在3.2 μmol/L吉非替尼中维持良好的生长状态,90% RPMI-1640+10% FBS继续培养48 h;Control组予以同步的二甲基亚砜(DMSO)处理;siLINC组采用Lipofectamine 2000介导LINC00628 小干扰RNA载体转染于GR细胞中;siControl组转染对照表达载体于GR细胞中;Blank组转染空白载体于GR细胞中;miR-145组转染miR-145过表达载体于GR细胞中;Rescue组共转染LINC00628过表达载体及miR-145过表达载体于GR细胞中,均使用1 μg/mL 嘌呤霉素筛选1周。
2 实时荧光定量PCR(qPCR) RNAiso Plus提取组织或细胞总RNA,取1 μg RNA进行逆转录,miR-145特异性逆转录引物序列,各待检测基因的合成引物序列如下(由上海生工提供):LINC00628:上游5′-AGAGCGAGCAGGATGAGATAGT-3′、下游5′-GTGAGCAAGGAAGTTGACAGTG-3′;miR-145:上游5′-CAGCATACATGATTCCTTGTA-3′、下游5′-CTTTGGTGTTTGAGATGTTTGG-3′; c-Myc:上游5′-CCTGGTGCTCCATGAGGAGAC-3′、下游5′-CAGACTCTGACCTTTTGCCAGG-3′; AKT1:上游5′-GGACAACCGCCATCCAGACT-3′、下游5′-GCCAGGGACACCTCCATCTC-3′;GAPDH:上游5′-GAAGGTGAAGGTCGGAGTC-3′ 、下游5′-GAAGATGGTGATGGGATTTC-3′。10 μL核酸荧光染料体系进行qPCR反应,每组设置3个平行孔,以GAPDH作为内参基因,2(-ΔΔCT)法计算LINC00628、miR-145、c-Myc及AKT1 mRNA的表达水平。
3 蛋白质印记技术(Western Blot) 收集细胞总蛋白,每组细胞取30 μg总蛋白进行10%聚丙烯酰胺凝胶电泳,稳压100V;转PVDF膜,稳流250 mA,5%脱脂牛奶室温封闭2 h,c-Myc、AKT1及GAPDH抗体1 ∶1 000孵育目的条带4℃过夜,1 ∶2 000二抗室温孵育1 h,Bio-rad ChemiDocXRS+化学发光仪进行曝光显影。
4 CCK-8实验 接种各组细胞于96孔细胞培养板,细胞浓度均为1 000个细胞/孔,设置0、0.25、0.5、1、2、4、8、16 μmol/L吉非替尼浓度梯度,每组每个梯度设置5个重复孔,培养48 h后加入CCK-8试剂,避光培养4 h,检测450 nm处吸光度,计算细胞吉非替尼半抑制浓度(Inhibitory concentration 50%,IC50)。
5 荧光素酶报告基因实验 连接LINC00628预测结合位点区域序列于psiCHECK2载体,培养293T细胞并分为2组:分别为miR-145 NC组及miR-145 mimics组,通过脂质体共转染miR-145 NC及psiCHECK2 LINC00628载体于miR-145 NC组细胞中,共转染miR-145 mimics及psiCHECK2 LINC00628载体于miR-145 mimics组细胞中,转染后48h后加入200 μL裂解液室温摇转孵育30 min,取10 μL加入96孔板,100 μL预混Luciferase Assay Reagent Ⅱ检测荧光强度为RLU1,100 μL预混Stop&Glo Reagent检测荧光强度为RLU2,以RLU1/RLU2作为各组细胞的相对荧光强度。
6 统计学方法 本研究使用SPSS 13.0进行统计学分析,单因素方差分析(One Way ANOVA)及LSD两两比较进行组间差异分析,检验水准α=0.05。
结 果
一、肺腺癌临床组织中LINC00628的表达情况
生物信息分析(数据平台:gepia.cancer-pku.cn/index.html)发现LINC00628在肺腺癌中高表达(图1A),同时高表达LINC00628的肺腺癌患者预后时间显著缩短(图1B),肺腺癌临床样本中同样发现LINC00628高表达(图1C)。
二、各组细胞中吉非替尼IC50的比较:CCK-8分析结果显示,Control组、GR组、siControl组及siLINC组,吉非替尼IC50分别为0.46±0.07、6.41±0.83、6.08±0.85及3.29±0.46 μmol/L,相比Control组,GR组、siControl组及siLINC组吉非替尼IC50均显著升高(P<0.05);相比siControl组,siLINC组吉非替尼IC50显著降低(P<0.05)(见图2A)。Blank组、miR-145组及Rescue组IC50分别为6.27±0.69、2.94±0.50及6.33±0.73 μmol/L,miR-145组吉非替尼IC50显著低于Blank组及Rescue组(P<0.05)(见图2B)。
三、各组细胞中LINC00628、miR-145、c-Myc及AKT1 mRNA的表达水平
qPCR结果显示,相比Control组,GR组LINC00628、c-Myc及AKT1 mRNA表达水平显著增高,miR-145表达水平显著降低(P<0.01);相比siControl组,siLINC组LINC00628、c-Myc及AKT1 mRNA表达水平显著降低,而miR-145表达水平显著增高(P<0.01)(见图3A);相比miR-145组,Blank及Rescue组miR-145表达显著降低,c-Myc及AKT1 mRNA表达显著增高(P<0.01)(见图3B)。

图1 肺腺癌临床组织中LINC00628的表达及其与患者预后的关系
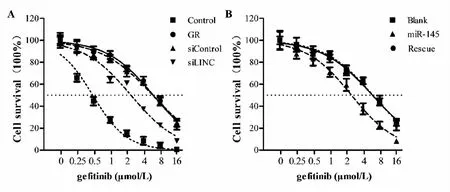
图2 各组细胞吉非替尼IC50的拟合曲线
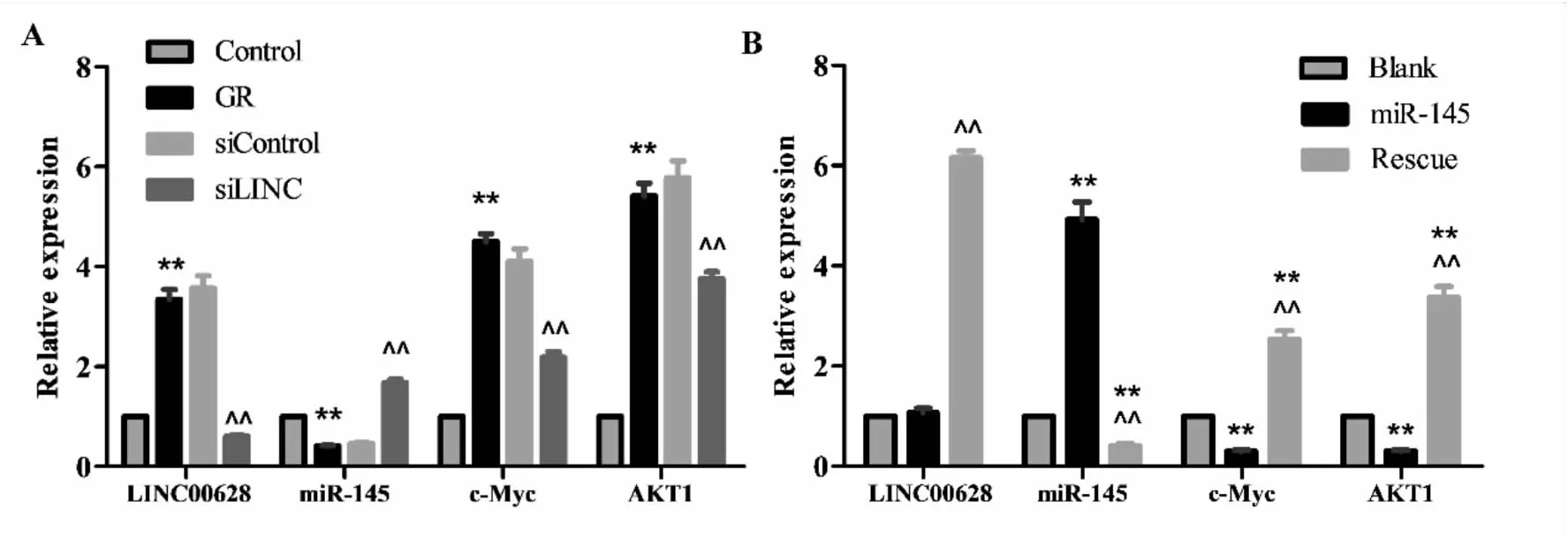
图3 各组细胞LINC00628、miR-145、c-Myc及AKT1 mRNA的表达及比较
四、各组细胞中c-Myc及AKT1蛋白表达水平
Western Blot结果显示,相比Control组,GR组c-Myc及AKT1蛋白表达水平显著增高;相比siControl组,siLINC组c-Myc及AKT1蛋白表达水平显著降低(见图4A、B)。相比miR-145组,Blank及Rescue组c-Myc及AKT1蛋白表达显著增高(见图4C、D)。
五、LINC00628与miR-145结合的验证
生物信息数据库(lncRNABase)预测发现LINC00628转录本715-721位点与miR-145碱基互补配对,通过构建LINC00628荧光素酶报告基因载体,发现miR-145 mimics可显著降低LINC00628-psiCHECK2的荧光信号(见图5)。

图4 各组细胞中c-Myc及AKT1蛋白印记
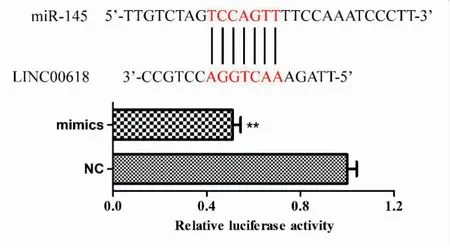
图5 LINC00628与miR-145结合位点的预测及验证
讨 论
miR-145的低表达与多种恶性肿瘤的发生及进展相关,包括卵巢癌、肝细胞癌、乳腺癌、结直肠癌及非小细胞肺癌等[3-5, 8-9],同时,有研究指出,在肺腺癌细胞中过表达miR-145后可使EGFR和NUDT1 mRNA表达显著下调,并抑制细胞增殖,另外miR-145在野生型或T790M点突变的EGFR肺癌细胞株中表达显著低于突变型EGFR细胞株,这提示miR-145的低表达与EGFR-TKI药物抵抗相关[10]。lncRNA作为机体表观遗传的重要执行者,通过竞争性结合miRNA,间接调控mRNA的表达,目前已发现LET、ROR、SNHG1、MALAT1等lncRNA参与调控miR-145,进而在恶性肿瘤中发挥生物学作用[11-13],但在肺癌中的相关调控机制仍有待探索。我们通过lncRNABase数据库在肺腺癌细胞模型中发现lncRNA LINC00628与miR-145存在可能的结合作用,同时GEPIA数据平台指出LINC00628在肺腺癌组织中高表达,并与患者预后不良相关,提示其在肺腺癌中的研究价值。
已有研究显示,LINC00628参与影响乳腺癌、胃癌、肝细胞癌及骨肉瘤细胞的恶性行为,D.Q. Chen等[14]研究显示,LINC00628在乳腺癌组织中低表达,并与患者预后时间正相关,体外过表达乳腺癌细胞系LINC00628可抑制细胞增殖及转移能力,并促进细胞凋亡,该效应与Caspase-3及Bax蛋白表达增加有关;Zi-Zhen Zhang等[15]发现LINC00628可促进组蛋白H3第27位赖氨酸上三甲基化(H3K27me3)水平,抑制胃癌细胞体内外增殖能力;另外,LINC00628也在肝细胞癌及骨肉瘤组织中低表达,并通过抑制VEGFA及PI3K/Akt通路在其中发挥抑癌效应[16-17]。这提示LINC00628与恶性肿瘤发生发展密切相关,但其与肺癌的关系仍不明确。因此,我们对其在肺腺癌中的表达情况及其与患者预后的关系进行了探究,GEPIA数据库及临床标本中发现LINC00628在肺腺癌组织中高表达,并观察到LINC00628的高表达预示患者预后不良。考虑到LINC00628与miR-145在肺腺癌细胞中存在潜在的相互作用以及miR-145与EGFR-TKI抵抗的密切关系,我们利用EGFR突变的肺腺癌细胞PC9作为体外模型构建了吉非替尼抵抗细胞株,发现GR细胞LINC00628与c-Myc及AKT1表达均上调,而miR-145表达显著降低,提示LINC00628可能参与肺腺癌细胞EGFR-TKI抵抗的形成。为了明确这一假设,我们在GR模型中,外源性干扰了LINC00628的表达,发现细胞miR-145表达升高,且c-Myc及AKT1表达降低,同时细胞吉非替尼敏感性增强,这表明干扰LINC00628可能通过促进miR-145的表达介导GR细胞吉非替尼抵抗的逆转。另外,我们在GR模型中过表达miR-145后发现c-Myc及AKT1表达降低,与文献中的结果趋势一致[3,5],且吉非替尼IC50下降,表明miR-145可抑制c-Myc及AKT1表达并促进细胞对吉非替尼敏感,而在共转染LINC00628及miR-145过表达载体后发现miR-145的效应被LINC00628所拮抗,提示两者可能存在调控作用。进一步的荧光素酶报告基因实验结果显示,LINC00628与miR-145相互存在结合作用。值得一提的是,LINC00628已被发现在多种恶性肿瘤中发挥抑癌作用[14-17],但本研究却揭示了其在肺腺癌中的促癌价值,这可能是由于不同来源的恶性肿瘤中主导肿瘤进展的分子机制不尽相同[18-19],而LINC00628可能存在包括原癌及抑癌基因在内的多种靶基因,导致其促癌作用及抑癌作用并存,而在肺腺癌中,其促癌作用显著大于抑癌作用,因此产生了与乳腺癌、胃癌、肝细胞癌及骨肉瘤中相反的生物学效应,其确切机制仍有待进一步探究。综上,我们的研究发现LINC00628可通过结合miR-145,影响下游基因c-Myc、AKT1的表达及细胞吉非替尼抵抗能力,是潜在的肺腺癌治疗靶点及分子标志物。
猜你喜欢
生物化学与生物物理进展(2022年5期)2022-05-23
电子乐园·上旬刊(2022年5期)2022-04-09
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2022年1期)2022-02-28
现代临床医学(2021年6期)2021-11-20
中老年保健(2021年5期)2021-08-24
山西医科大学学报(2021年7期)2021-08-07
小雪花·成长指南(2021年2期)2021-05-20
中国药学药品知识仓库(2021年18期)2021-02-28
初中生世界·九年级(2019年4期)2019-05-05
