
利用CRISPR/Cas9 系统构建可诱导型猪Sohlh1 基因敲除细胞系
2021-03-02 04:57:38乔延召李清华黄建豪卫恒习张守全
华南农业大学学报 2021年2期
乔延召,刘 凯,李清华,黄建豪,卫恒习,张守全
(1 华南农业大学 动物科学学院/国家生猪种业工程技术研究中心,广东 广州 510642; 2 汕尾宝山猪场有限公司,广东 汕尾 516600)
精卵发生特异碱性螺旋环螺旋转录因子(Spermatogenesis and oogenesis specific basic helixloop-helix transcription factor,Sohlh1)是一种主要在睾丸和卵巢中表达的组织特异性基因,能够调控精子和卵子的发生[1]。公鼠睾丸中Sohlh1的mRNA最早可在胚胎发育12.5 d 时检测到,在胚胎发育15.5 d 时通过免疫组化检测到了SOHLH1 蛋白,免疫荧光试验进一步证实SOHLH1 蛋白在精原细胞中的特异性表达[1]。母鼠卵巢中Sohlh1的mRNA最早可在胚胎发育13.5 d 时卵细胞进入减数分裂Ⅰ期检测到,蛋白则在胚胎发育15.5 d 时检测到,在卵母细胞中SOHLH1 蛋白同时存在于细胞核和细胞质中[2]。研究表明,Sohlh1可能被视为原发性卵巢功能不全的调控基因[3],非梗阻性无精子症也与Sohlh1基因的突变有关[4]。Sohlh1在生殖发育中更多的调控机制有待于进一步研究,以往的研究显示Sohoh1作为生殖细胞特异表达基因,其启动子被广泛应用于生殖细胞特异表达启动子,敲除此基因则被广泛应用到生殖发育障碍动物模型中。
作为一种理想的基因编辑技术,CRISPR/Cas9技术在模式动物制备[5-6]、遗传疾病治疗[7-9]、癌症研究[10-11]等多个领域得到了广泛的应用。但CRISPR/Cas9 系统存在脱靶问题[12],效率受影响,Slaymaker等[13]把3 个带正电荷的氨基酸突变为非极性的氨基酸,大大降低了CRISPR/Cas9 系统的“脱靶”切割,使其效率显著提高。除基因编辑技术外,慢病毒转基因法以其稳定、高效整合的优势,也常常被应用在转基因细胞和动物生产中。
四环素诱导表达系统因高效、可控和低渗漏等优点深受研究者喜爱。四环素调控系统中的Tet-on系统又称四环素开启系统,指只有在诱导剂四环素或其衍生物强力霉素存在的条件下,rtTA 蛋白才能结合到Tet 应答元件(Tet-responsive element,TRE)序列上产生激活作用,使插入的目的基因进行表达[14-18]。这种安全可调控的特点极大地方便了目的基因多方面的研究[19]。利用Tet-on 系统能够对目的基因时空表达进行调控的特点,本研究将CRISPR/Cas9 系统与Tet-on 系统相结合,通过条件性地对Sohlh1基因进行敲除,达到构建可诱导型Sohlh1基因敲除细胞系的目的,以此细胞系建立的克隆猪生殖模型,将有助于Sohlh1基因功能、生殖细胞发育以及人类生殖障碍等问题的研究。
1 材料与方法
1.1 供试材料
1.1.1 试验动物组织 仔猪睾丸组织和30 d 胎龄猪胎儿采自广东温氏集团水台原种猪场。
1.1.2 质粒和细胞株 pLV-sgRNA(#50662)质粒、pGL3-GFP 质粒购于Addgene 公司;低脱靶优化慢病毒载体PCW-eSpCas9(1.1)(含Tet-on 系统)、稳转Cas9 蛋白猪胎儿成纤维细胞(Porcine fetal fibroblast,PFF)系、慢病毒包装质粒psPAX2、pMD2.G 由国家生猪种业工程技术研究中心保存;293FT 细胞株购于ATCC 公司。
1.1.3 主要试剂 pMD™18-T Vector Cloning Kit 购自TaKaRa 公司;感受态细胞Escherichia coliStbl3 和DH5α、TransScript II One-Step gDNA Removal and cDNA Synthesis SuperMix(AT311)、质粒提取试剂盒购自全式金生物公司;SpeⅠ、EcoRⅠ、XhoⅠ、NheⅠ等限制性内切酶购于Thermo 公司;ClonExpress® MultiS One Step Cloning Kit 购自南京诺唯赞公司;GML-PC 慢病毒浓缩试剂盒、HGTrans293™transfection reagent、LipofectamineTM2000、Attractene Transfection Reagent 购自上海吉满生物公司;灭稻瘟菌素、嘌呤霉素购自Sigma 公司;动物组织总RNA 提取试剂盒、细胞基因组提取试剂盒购自天根生化公司;凝胶回收试剂盒购自Magen公司;全蛋白提取试剂盒购自凯基公司。
1.2 方法
1.2.1 PFF 细胞的获取 取纯种杜洛克公猪30 d胎龄的胎儿,用含有双抗的PBS 缓冲液清洗,75%(φ)乙醇溶液消毒全身,在超净台内去除头、尾、四肢、心脏及其他脏器,双抗液再次清洗后,于培养皿中用眼科剪将胚胎充分剪碎,使组织块直径小于1 mm;随后加入37 ℃预热的DMEM/F12 培养液,转移混合液至卡式瓶,混合均匀并放入培养箱中培养,5 h后再加入适量含20%(φ)FBS 的DMEM/F12 培养液,24~48 h 后观察组织块旁细胞生长状况。待长出的 PFF 细胞达到足够密度时,进行细胞冻存。
1.2.2 PCW-eSpCas9(1.1)慢病毒载体侵染PFF 细胞及检测 复苏保存的PFF 细胞并进行培养,待细胞汇合度(细胞生长面积/孔板面积)达约80%时,用PCW-eSpCas9(1.1)慢病毒载体浓缩液侵染并放回培养箱中培养,12 h 后换液为新鲜培养基继续培养。病毒侵染2 d 后换用含有1 μg/mL 嘌呤霉素的完全培养基继续培养,每天换液,筛选大约2 周,待细胞稳定生长,一部分进行冻存;另外一部分加入强力霉素(Dox)进行诱导,最后收集诱导组、未诱导组细胞进行RT-PCR 和Western-blot 检测。
1.2.3 pLV-sgRNA 载体的改造及验证 以pLVsgRNA(#50662)质粒和pGL3-GFP 质粒为模板,融合PCR 得到2A-GFP 片段。再用SpeⅠ和EcoRⅠ双酶切pLV-sgRNA 质粒,得其线性化片段。随后将2A-GFP 片段与pLV-sgRNA 线性化片段进行连接(步骤见One Step Cloning Kit 连接试剂盒说明书),产物转化感受态细胞Stbl3,挑取单菌落至含有氨苄青霉素的LB 培养液中培养12 h,菌液PCR 鉴定,反应程序为:94 ℃预变性5 min;94 ℃变性30 s、60 ℃退火30 s、72 ℃延伸1 min,32 个循环;72 ℃延伸10 min。将阳性质粒送生工测序。最后根据LipofectamineTM2000 转染手册将测序正确的改造载体转入293FT 细胞进行荧光检测。
1.2.4 猪Sohlh1基因的扩增及靶位点预测 依据动物组织总RNA 提取试剂盒说明书抽提仔猪睾丸组织RNA,按反转录试剂盒要求进行反转录;根据NCBI 数据库记录的猪Sohlh1基因序列,使用Primer Premier 5.0 软件设计Sohlh1扩增引物(Sohlh1-F:ATGGCGTCCGGGGCTCCCGAG;Sohlh1-R:TCAGTAGGCAAAGAAGTCAG),以cDNA 为模板,进行Sohlh1基因的PCR 扩增,胶回收DNA 片段进行T-A 克隆、连接pMD™18-T 载体、转化DH5α 感受态细胞、菌液PCR 鉴定,选取阳性菌测序,将结果在NCBI 数据库中进行比对。最后把测序所得猪Sohlh1基因CDs 区输入sgRNA Designer 软件(http://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design),根据评分和所在外显子区域选出合适的靶基因位点。
1.2.5 pLV-sgRNA-2A-GFP 特异性载体的构建及慢病毒包装 以PCR 扩增的人U6 启动子片段和含有靶位点的sgRNA 片段为模板,融合PCR 得到U6-sgRNA 瞬时表达载体,用无血清培养基稀释后,加入转染试剂Attractene Transfection Reagent,形成核酸−转染试剂复合物。将复合物转染已筛选并验证表达Cas9 蛋白的PFF 细胞,于48~72 h 收集细胞进行基因组提取及PCR 扩增,引物为F-S3gS4g、R-S5gS6g(表1),随后对敲除基因片段进行分子克隆操作(同“1.2.4”),测序鉴定,进行敲除活性分析。根据Sohlh1基因靶位点敲除分析结果,选取目标靶点构建pLV-sgRNA-2A-GFP 特异性载体:用XhoⅠ和NheⅠ酶切已构建好的pLV-sgRNA-2AGFP 质粒,得其线性化载体并与U6-sgRNA 瞬时表达载体进行连接,产物转化Stabl3 感受态细胞,分子克隆步骤同“1.2.4”,阳性质粒送生工测序比对。特异性载体的慢病毒包装检测:用Opti-DMEM稀释混合好的DNA(穿梭质粒、psPAX2 和pMD2.G质量比为2∶2∶1),加入转染试剂HG-Trans293TM transfection reagent,然后转入293FT 细胞中,于37 ℃、CO2体积分数为5%的培养箱内培养,分别在48 和72 h 后收集病毒上清液,用GML-PC 慢病毒浓缩试剂盒进行浓缩。用1 mL 浓缩液和1 μL 终浓度为10 μg/mL 的Polybrene共同侵染293FT 细胞,48 h 后显微镜观察荧光表达情况。

表 1 Sohlh1 基因敲除鉴定引物Table 1 Primers for Sohlh1 gene knockout identification
1.2.6 筛选细胞系阳性比例鉴定 用上述pLV
sgRNA-2A-GFP 特异性慢病毒载体侵染稳转PCWeSpCas9(1.1)基因PFF细胞(图1),3~4 d 后换用含3μg/mL 的灭稻瘟菌素、15%(φ)血清的新鲜培养基培养。每2~3 d 用含有抗生素的新鲜培养基换液并随时观察细胞荧光表达情况,一段时间以后待大部分细胞都有荧光表达时,停止药物筛选。培养药物筛选的PFF 阳性细胞至80%的细胞汇合度,更换含有Dox 的完全培养液继续培养,3 d 后收集细胞,抽提基因组,进行PCR 扩增,引物为F-S3gS4g、R-S3gS4g(表1),分子克隆步骤同“1.2.4”,最后将阳性菌送生工测序并统计样品敲除效率。
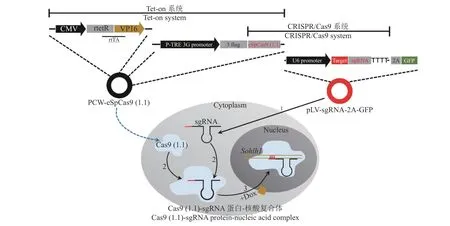
图1 sgRNA 与Sohlh1 基因靶向结合示意图Fig.1 Schematic diagram of targeted binding of sgRNA and Sohlh1 gene

图2 优化后的低脱靶慢病毒载体PCW-eSp Cas9(1.1)结构Fig.2 Structure of optimized low miss lentivirus vector PCW-eSp Cas9(1.1)
2 结果与分析
2.1 转PCW-eSpCas9(1.1 )基因细胞系的检测
慢病毒包装质粒PCW-eSpCas9(1.1)(图2)侵染PFF细胞,嘌呤霉素筛选得到细胞克隆团(图3)。Dox 诱导转PCW-eSpCas9(1.1)基因PFF细胞系后进行RT-PCR 检测,结果显示eSpCas9(1.1)基因只在诱导后的细胞中转录(图4)。Western-blot 检测结果显示Dox 诱导组细胞能够表达eSp Cas9(1.1)蛋白,未诱导组细胞不表达该蛋白(图5)。综合以上结果,可以得出eSpCas9(1.1)基因已整合到PFF细胞基因组上并稳定表达。
2.2 pLV-sgRNA 载体的改造结果
融合PCR 得到的2A-GFP片段与酶切p LVsgRNA 质粒后得到的片段(图6)进行连接、转化,阳性菌测序正确。重组质粒转染293FT 细胞,转染后48 h 荧光显微镜观察到带绿色荧光的细胞(图7),表明pLV-sgRNA 载体成功地改造为pLV-sgRNA-2A-GFP 载体。

图3 嘌呤霉素筛选的PCW-eSp Cas9(1.1)基因阳性PFF细胞克隆团Fig.3 PCW-eSpCas9(1.1)gene positive PFF cell clones screened by purinomycin
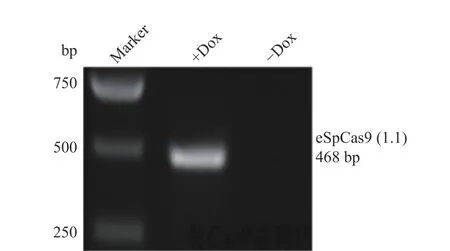
图4 RT-PCR 检测表达eSp Cas9(1.1)基因细胞系Fig.4 Detection of cell line expressing eSpCas9(1.1)gene by RT-PCR

图5 Western-blot 检测表达eSpCas9(1.1)蛋白细胞系Fig.5 Detection of cell line expr essing eSp Cas9(1.1)protein by Western-blot

图6 融合PCR 扩增得到2A-GFP 片段(A)和双酶切p LV-sgRNA 载体得到的线性化片段(B)Fig.6 2A-GFPfragment obtained by fusion PCR (A)and the linearized fragment obtained by doubleenzyme digestion of p LV-sgRNA vector (B)

图7 荧光显微镜检测p LV-sgRNA-2A-GFP质粒转染后的293FT细胞Fig.7 Detection of 293FT cells transfected with the p LVsgRNA-2A-GFP plasmid by fluorescence microscopy
2.3 猪Sohlh1基因测序及靶位点的选择
Lasergene软件比对PCR 扩增后测序所得猪Sohlh1基因与NCBI 数据库所报道的猪Sohlh1基因,结果表明两者序列基本一致,仅在起始密码子下游894 bp处有1个碱基不同(图8),但是所编码的均为谷氨酸,对Sohlh1功能无影响。再通过软件预测Sohlh1基因CDs区全部的sgRNA 位点,分析各位点的评分及所在的外显子区域,从中选择符合条件的4个靶位点(表2)。

图8 PCR 扩增猪Sohlh1 基因与NCBI 数据库对比Fig.8 Comparison of pig Sohlh1 gene amplified by PCR with NCBIdatabase
2.4 p LV-sgRNA-2A-GFP特异性载体的构建及慢病毒包装检测
PCR 扩增出U6启动子与含有靶位点的sgRNA片段(图9A),融合PCR 得到U6-sgRNA 瞬时表达载体(图9B)。为验证敲除效果,使用U6-sgRNA 转染转Cas9基因PFF细胞,转染后48~72 h 抽提基因组进行PCR 扩增,得到1 593 bp的敲除基因片段,经克隆、测序得到各靶位点的敲除情况(图10)。试验送样72个,有效结果中3、4、6号位点突变的样品数分别为17、16及14个,3、4号位点同时突变的为8个,5号位点均无突变,分析测序结果知3、4、6号靶位点有敲除活性,同时3、4号位点可形成同时断裂的切割,利于提高突变几率(图10)。XhoI与NheI双酶切pLV-sgRNA-2A-GFP质粒,产物大小为7807 bp(图9C),与带有3、4号靶位点的U6-sgRNA 载体连接,阳性菌测序正确。将pLV-sgRNA-2A-GFP特异性载体进行慢病毒包装,其浓缩液侵染293FT细胞,48 h 后观察到293FT细胞的荧光表达,表明pLV-sgRNA-2A-GFP特异性载体慢病毒包装成功(图11)。

表 2 Sohlh1基因靶点的预测与评分Table 2 Prediction and scoring of Sohlh1 gene targets
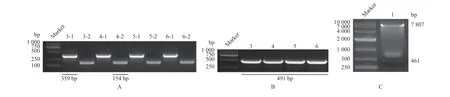
图9 PCR 检测p LV-sgRNA-2A-GFP 特异性载体的构建Fig.9 Construction of p LV-sgRNA-2A-GFP specific vector and PCR examination

图10 不同靶位点敲除的序列分析Fig.10 Sequence analysis of different target knockout

图11 特异性载体慢病毒浓缩液侵染293FT细胞荧光显微镜观察Fig.11 Fluorescence microscopy observation of 293FT cells infected by sp ecific vector lentivir us concentrate
2.5 慢病毒侵染PFF 细胞及阳性细胞筛选结果
稳转PCW-eSp Cas9(1.1)基因PFF细胞进行pLV-sgRNA-2A-GFP特异性慢病毒载体侵染,灭稻瘟菌素筛选3 d 左右细胞大量死亡。图12为药物筛选7、14、28 d 荧光显微镜观察结果,可以看到28 d所筛选的细胞系有约90%以上的细胞表达了绿色荧光蛋白,表明此时pLV-sgRNA-2A-GFP特异性慢病毒载体侵染PFF细胞系筛选成功。在筛选成功的PFF细胞系中加入Dox 诱导培养72 h,PCR 扩增Sohlh1基因含有3、4号靶位点的片段,经分子克隆与测序,结果显示测序的20个菌液中,3号位点有突变的样品数为14个,突变率为70%,4号位点有突变的样品数为7个,突变率为35%,而3、4号位点同时发生移码突变的样品数为4个,突变率为20%,最终测得样品突变总数为17个,有敲除的样品比例达到85%。
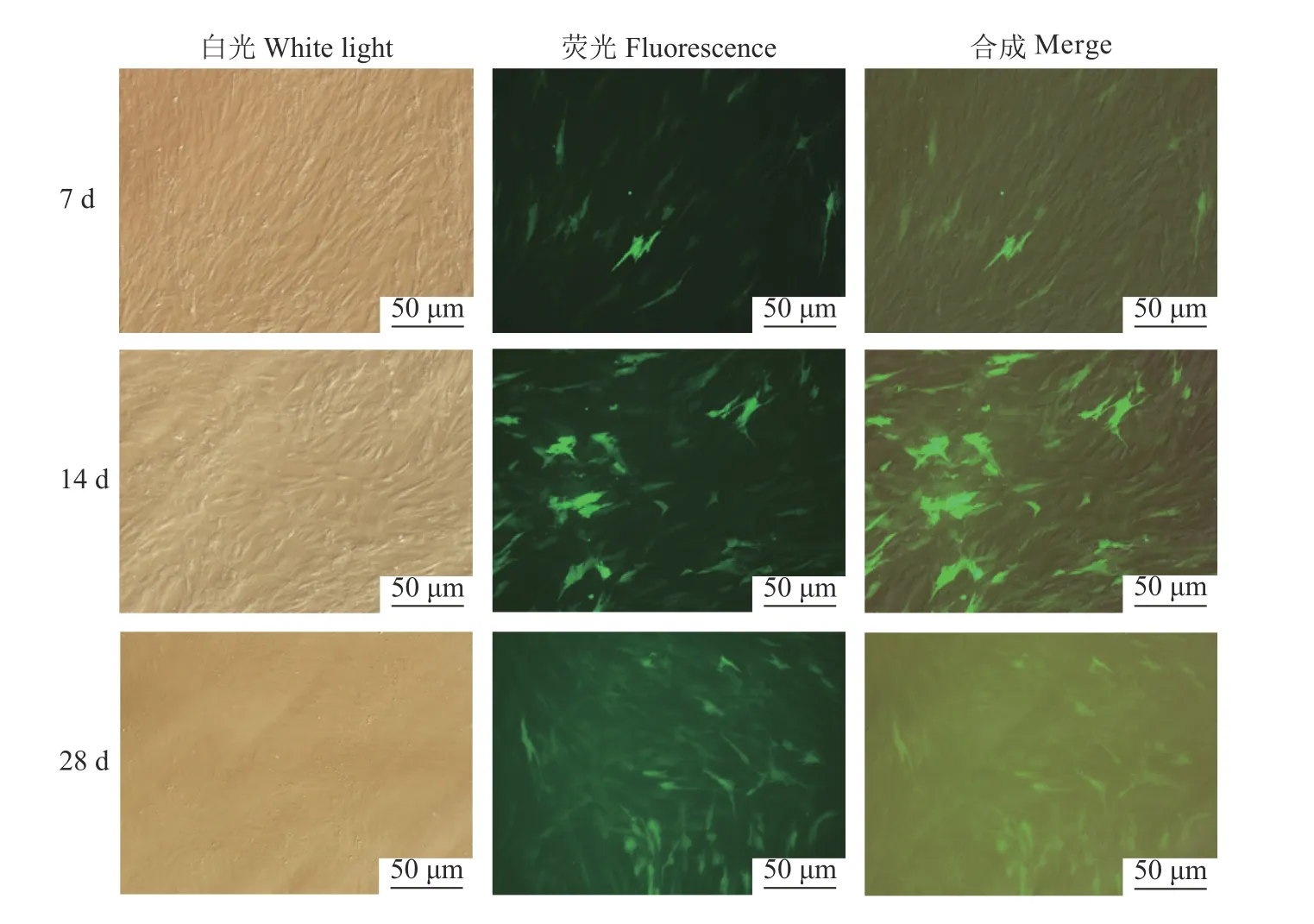
图12 药物筛选不同天数荧光显微镜观察Fig.12 Fluorescence microscopy observation on different days of drug screening
3 讨论与结论
生殖细胞特异性基因由于只在生殖细胞中表达,对于发育生物学研究显得十分重要。基因敲除试验证明Sohlh1基因在生殖细胞成熟过程中起着重要的作用[1-2],成年Sohlh1(−/−)雌鼠卵巢的临床表型与人类卵巢早衰(Premature ovarian failure,POF)患者相似,而且雄性性腺机能衰退[20]等也与Sohlh1基因的突变有关。本研究选择Sohlh1为靶基因,通过软件预测Sohlh1基因CDs区全部的sgRNA 位点,构建U6-sgRNA 瞬时表达元件进行敲除效果的验证,测序得到各靶位点的敲除情况,成功地筛选到有敲除活性的靶位点,为研究敲除后Sohlh1基因的功能打下了基础。
本研究使用的Tet-on 系统,它的调控元件为反式应答转录激活因子(Reverse tTA,rtTA),在不加入诱导剂Dox 时,rtTA 不与表达目的基因的Tet 应答元件(TRE)结合,无法启动目的基因表达,当加入Dox 诱导后,rtTA 结构改变,能够结合到TRE上,激活插入的目的基因表达。本研究利用已经优化好的低脱靶率慢病毒载体PCW-eSpCas9(1.1)(含Tet-on 系统),通过Tet-on 系统与CRISPR/Cas9系统的紧密结合,使用构建好的p LV-sgRNA-2AGFP特异性慢病毒载体侵染含有上述系统的PFF细胞,由Tet-on 系统调控Cas9基因的表达,在试验中加入Dox 诱导表达Cas9蛋白对Sohlh1基因进行切割,造成Sohlh1基因突变,测序结果显示有敲除样品的比例高达85%,成功地构建了可诱导型猪Sohlh1基因敲除细胞系。
猪在生理结构上与人类极为相似,包括胃肠道、肾脏、外神经、皮肤、内分泌系统、生殖系统等[21],因此,猪可作为一种重要的大动物模型应用于预防和治疗药物的临床前试验[22]、人类疾病发病机制及功能基因组学研究[23-24]。但猪制备生殖疾病模型鲜有报道。本研究构建的敲除Sohlh1基因细胞系可用于后期条件性生殖细胞敲除猪模型的制备,为后期研究Sohlh1基因的功能和解决人类生殖疾病相关问题创造条件。
本研究通过改造和优化的慢病毒载体介导,利用Tet-on 系统和CRISPR/Cas9系统成功制备了可诱导型Sohlh1基因敲除PFF细胞系,为研究Sohlh1基因的功能以及制备条件性敲除Sohlh1基因生殖模型猪储备了试验材料。
猜你喜欢
生物技术通报(2023年2期)2023-03-07 12:54:58
当代水产(2022年1期)2022-04-26 14:35:30
西南农业学报(2016年5期)2016-05-17 05:42:33
西南农业学报(2016年6期)2016-04-16 05:12:51
山东医药(2015年14期)2016-01-12 00:39:43
江苏大学学报(医学版)(2015年2期)2015-04-17 06:49:51
中国医药导报(2015年26期)2015-02-28 22:07:44
中国药业(2014年21期)2014-05-26 08:56:45
中华介入放射学电子杂志(2014年1期)2014-02-02 05:24:06
现代检验医学杂志(2014年6期)2014-02-02 03:01:54
