
Gastrointestinal tumors and infectious agents: A wide field to explore
2021-02-12 07:19MiriampezmezBelGarcdeSantiagoPedroDavidDelgadopezEduardoMalmiercaJesGonzlezOlmedosarmezRaposoCarmenSandovalPilarRuizSecoNoraEscribanoJorgeFranciscomezCerezoEnriqueCasado
World Journal of Meta-Analysis 2021年6期
Miriam López-Gómez, Belén García de Santiago, Pedro-David Delgado-López, Eduardo Malmierca, Jesús González-Olmedo, César Gómez-Raposo, Carmen Sandoval, Pilar Ruiz-Seco, Nora Escribano, Jorge Francisco Gómez-Cerezo, Enrique Casado
Miriam López-Gómez, Medical Oncology Department.Precision Oncology Laboratory, Infanta Sofía University Hospital, San Sebastián de los Reyes 28231, Madrid, Spain
Belén García de Santiago, Pharmacy Department, Infanta Sofia University Hospital, San Sebastián de los Reyes 28703, Madrid, Spain
Pedro-David Delgado-López, Neurosurgery Department, Burgos University Hospital, Burgos 09006, Spain
Eduardo Malmierca, Pilar Ruiz-Seco, Jorge Francisco Gómez-Cerezo, Internal Medicine Department, Infanta Sofía University Hospital, San Sebastián de los Reyes 28703, Madrid, Spain
Jesús González-Olmedo, César Gómez-Raposo, Carmen Sandoval, Enrique Casado, Medical Oncology Department, Infanta Sofia University Hospital, San Sebastián de los Reyes 28703, Madrid, Spain
Nora Escribano, Intensive Care Unit, Jiménez Díaz Foundation, Madrid 28040, Madrid, Spain
Abstract Infection is currently one of the main contributors to carcinogenesis.In fact, the International Agency for Research on Cancer has categorized eleven biological agents as group I carcinogens.It is estimated that around 16% of the 12.7 million new cancers diagnosed in 2008 were attributable to infectious agents.Although underdeveloped regions carry the highest incidence rates, about 7.4% of infectionrelated cancer cases occur in developed areas.Physicians are increasingly aware of the potential carcinogenic role of common virus like the Human Papilloma virus in cervical cancer, or the hepatitis B and C viruses in hepatocarcinoma.However, the carcinogenic role of several other infectious agents is less recognized.Given that gastrointestinal malignancies carry an overall poor prognosis, a better understanding of the carcinogenic mechanisms triggered by infectious agents is key to decrease the rate of cancer related deaths.Preventive measures directed to such infections would ideally impact survival.In this paper we review the main pathogenic mechanisms related to the development of gastrointestinal malignancies induced by infectious microorganisms and other pathogens which are currently under investigation.
Key Words: Gastrointestinal tumors; Infectious agents; Bacteria; Virus; Prevention
INTRODUCTION
During the last decades the causal relation between several pathogens and the onset of cancer has been firmly established[1].However, the relationship between pathogens and cancer has been subject of research since the end of the XIX century.Rous[2,3] (1879-1970) discovered that sarcomas affecting domestic fowls could be transferred to other fowls through a viral vector, later known as Rous sarcoma virus (RSV).This finding was awarded with the Nobel Prize in 1966 and demonstrated for the first time in history that a malignant tumor could be induced by an infectious agent.In fact, the RSV was considered the first oncogenic retrovirus.Subsequently, other tumorinducing viruses affecting animals were identified[4-6].In 1964, the first oncogenic virus affecting humans, the Epstein Barr virus, was identified[7].
At present, a great effort is being done in order to elucidate the underlying connection between infectious agents and cancer.In 2018, an estimated 2.2 million infection-attributable cancer cases were diagnosed worldwide, corresponding to an age-standardized incidence rate (ASIR) of 25.0 per 100.000 person-years[8].Currently, 11 infectious agents have been classified as Group 1 Carcinogens by the International Agency for Research on Cancer (IARC)[1].Helicobacter pylori(H.pylori) (810.000 cases, ASIR 8.7), Human Papilloma virus (690.000, 8.0), Hepatitis B virus (360.000, 4.1) and hepatitis C virus (HCV) (160.000, 1.7) comprise the primary causes of infectious-related tumors, accounting for more than 90% of infection-related cancers worldwide[9].
VIRAL INFECTIONS
Viral chronic infection can promote cancerviathree different pathogenic mechanisms:
(1) Induction of chronic inflammation provoked by the immune response to a persistent infection; HCV-related liver cancer is a paradigmatic example, in which persistence of viral replication within the liver parenchyma maintains a state of local chronic inflammation involved in cancer development[10]; (2) Virus-induced genome transformation secondary to viral persistence in infected cells; This mechanism is typically found in Epstein-Barr virus (EBV) associated Burkitt’s lymphoma.EBV replicating in the oral epithelium can transform resting B lymphocytes into proliferating lymphoblastoid cell lines involved in oncogenesis[11]; and (3) Promotion of chronic suppression of host immune response.Patients infected with the human immunodeficiency virus can be deeply immune-suppressed.These individuals exhibit an increased predisposition to develop infection-driven tumors as the mechanisms of immunosurveillance are disrupted by the viral replication[12].
Although these mechanisms may occur simultaneously[13], some other yet unknown need also be involved, since many other pathogens inducing similar immune alterations do not seem to induce cancer.It is widely recognized that viruses can cause cellular malignant transformation by inducing genetic instability.In fact, viruses are able to fuse their genome with that of the host resulting in activation of multiple oncogenes and intracellular signaling pathways leading to cellular proliferation, inflammation and immune dysregulation[14,15].
BACTERIAL INFECTIONS
Recent studies have shown that certain infectious agents like molds, helminths and bacteria, capable of interacting with mammalian host cells, can induce cancer, even in the absence of genomic alteration[16].Unlike the well-established relation between viruses and carcinogenesis, the oncogenic role of bacterial infection remains controversial[17].The molecular mechanisms by which bacteria might promote carcinogenesis are currently under research[10], and both bacterial and host factors seem to be involved.Bacterial cell-surface components, toxins and effector proteins can interact with host cells by activating essential signaling pathways involved in cancer formation[16].
Role of bacterial surface antigens in carcinogenesis
The bacterial surface exhibits several antigens that interact with the host and activate both innate and adaptative immune responses, allowing them to escape the host immune system.For example, gram-negative bacteria can coat their outer surface with a polysaccharide capsule, thus preventing the activation of the Complement cascade and the phagocytic process[18].Other bacteria are able to modify some surface molecules, like lypo-polysaccharides, flagella and peptidoglycans, to avoid immune recognition[19].Additionally, some other bacteria can express a variety of surface proteins that facilitate attachment to host cells and subsequent internalization[20].These strategies aim to improve bacterial survival by eluding the immune response.However, definitive cellular malignant transformation needs to be induced by specific intracellular signaling cascade activation driven by certain bacterial molecules.For example,H.pylorican directly activate the RAS/mitogen activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway resulting in malignant transformationviathe CagL protein (an adhesin molecule) that binds to the beta5-integrin expressed by host cells[21];H.pylorialso expresses the OipA protein that binds to the EGFR host receptor resulting in AKT and beta-catenin signaling activation, which also promotes carcinogenesis[22].
Immune cell elimination
To ensure survival among host cells, pathogenic bacteria have developed several strategies to attack the immune system.For example, they can secrete cytolytic toxins through their outer membranes that can affect the endosomal system.Bacterial toxins may induce carcinogenesis altering the host cell environment by inducing genome instability, promoting resistance to cell death signaling, and enhancing proliferative signaling[23].
Induction of genome instability:Bacterial toxins are capable of inducing host cell damage in the DNA double helix.Cytolethal Distending Toxin, Collibactin, Shiga toxin and endonucleases are some examples[24].DNA damage causes immune host cells to arrest the cell cycle at the G1-S or G1-M stages.
Resistance to cell death signaling and induction of proliferative signaling:Toxins produced by some pathogens, likeBacteroides fragilis, can bind to intestinal epithelial cell receptors and induce cellular proliferation and differentiation.Bacteroides fragiliscan silence the tumor suppressor protein E-Cadherin, resulting in the activation of beta-catenin/Wnt and nuclear factor kbeta (NF-kB) signaling pathway[25].
Host cells transformation secondary to bacterial proteins
To induce malignant transformation, bacteria must ensure a persistent infection within the cells of the new organism.They need to be internalized, must control their own growth and facilitate their release.Usual non-pathogenic bacteria are typically phagocyted in the phagosome, and then merged with the lysosome and turned into a phagolysosome, where they are finally destroyed.However, some bacteria have developed a number of mechanisms that avoid the formation of the phagolysosome, allowing them to freely enter the cytosol[26]; some bacteria, for example, secrete proteins that induce pore formation in the organelle[27]; while others have developed different mechanisms to avoid destruction inside the phagolysosome[28].
The ultimate explanation by which bacteria promote cancer and whether they obtain any survival advantage remains unknown.This is an interesting and fastgrowing research field, in which prevention measures directed to infection may impact overall cancer development.The oncogenic role of various infectious agents regarding gastrointestinal tract malignancies are now described.Since these tumors carry relevant mortality rates, screening programs directed to identify such pathogens may be beneficial.
ESOPHAGEAL CANCER
The incidence of esophageal tumors differs substantially across countries.The highest rates are found in Southern and Eastern Africa and Eastern Asia.Although the histopathological squamous variant has been associated to several viral subtypes, the pathogenic role of viruses in the development of adenocarcinoma is yet unclear.
Human papilloma virus
HPV family viruses are non-enveloped DNA tumor viruses transmitted by sexual contact.More than 100 subtypes of HPV have been described, of which at least 14 carry a significantly increased risk of tumor development [29].HPV infects epithelial cells and is able to integrate itself inside the host genome.Some oncoproteins produced by the virus (mainly E6 and E7) alter tumor suppressor pathways and promote malignant proliferation[30].In 1982, Syrjänenet al[31] observed the typical morphological lesions present in HPV related-condylomas in both benign and cancerous esophageal tissues.These findings supported the hypothesis that HPV was involved in the pathogenesis of esophageal malignancies.Subsequent immunohistochemical studies demonstrated that HPV structural proteins were present in malignant lesions of patients from different continents[32].In fact, the oncogenic potential role of HPV in the development of squamous esophageal carcinoma has been the subject of several metanalysis[33-35].The first metanalysis of case-control studies investigating the role of HPV in esophageal tumors was conducted in 2013 by Liyanageet al[36].They gathered 21 studies, including 1223 patients and 1415 controls.The authors calculated a pooled OR for HPV and squamous esophageal tumors of 3.04 [95% confidence interval (CI): 2.20 to 4.20].Noticeably, countries with a low to medium esophageal cancer incidence showed a stronger relationship (OR 4.65, 95%CI: 2.47 to 8.76) than regions with higher incidence (OR 2.65, 95%CI: 1.80 to 3.91).The authors concluded that HPV infection was associated with a 3-fold risk of squamous esophageal cancer and made an urgent call to the IARC to promote the use of vaccines against HPV in high-risk populations.
In 2016, Wanget al[37] published a systematic review and meta-analysis on the association between HPV 16 and 18 subtypes and esophageal cancer worldwide.They included 32 studies and showed that HPV infection rate in the tumoral cohort was 46.5%vs26.2% in controls (OR = 1.62; 95%CI: 1.33-1.98), providing strong epidemiologic evidence supporting the association between HPV infection and esophageal tumors[37].
EBV
EBV is an oncogenic virus that belongs to the gamma-herpes virus family.It is a widespread pathogen carried by 95% of the population worldwide.Fortunately, the majority of EBV infections are subclinical[38].Although, the EBV typically infects B lymphocytes, recent research has shown that it can also infect epithelial cells, and suggests a potential association with esophageal carcinoma, yet to be confirmed.Jenkinset al[39] detected for the first time EBV in esophageal tumors, however, only in 5 out of 60 tumors.Awerkiewet at[40] demonstrated the presence of EBV-DNA in 35% of squamous cell carcinomas and 36% of adenocarcinomas, using nested polymerase chain reaction (PCR) diagnostic techniques.
In areas with higher incidence of esophageal carcinoma, the association between EBV and esophageal cancer has been investigated.Wuet al[41] studied the coinfection of EBV and Herpes Simplex virus (HSV) and detected, by in situ hybridization and immunohistochemistry, the EBER and LMP-1 proteins in 6.1% of carcinoma specimens, preferentially among poorly differentiated squamous cell carcinomas and undifferentiated carcinomas with intense lymphoid infiltration.Other authors have contributed to elucidate the role of EBV in the carcinogenesis of esophageal carcinoma[42,43].In summary, EBV infection has been related to the etiology of some variants of esophageal carcinoma, at least in countries with increased prevalence.However, more studies are needed to establish the exact pathogenic role of EBV in esophageal carcinogenesis.
Herpes simplex and Citomegalovirus
HSV-1 infection occurs mainly in the mouth and has been related to oral cancer[44].Citomegalovirus (CMV), a common human pathogen has been associated with cervical and non-melanoma skin cancer[45,46].Zhanget al[42] reported an increased predisposition of esophageal carcinoma in patients infected with HSV-1 (OR 10.3 with 95%CI: 3.331.6).HSV infection is primarily related to esophagitis, which usually precedes esophageal carcinoma.However, no CMV was detected in any of the samples[42].Wuet at[41] found HSV DNA and HSVI, II protein expression in 52 (31.7%) of the 164 tumors analyzed supporting the relationship between HSV and esophageal carcinoma cell differentiation with lymphocyte infiltration in the tumoral stroma.However, other authors have failed to demonstrate the relation between HSV/CMV and esophageal carcinoma, therefore leaving the question open[47].
GASTRIC CANCER
H.pylori
Gastric cancer can be generally classified in cardia (upper stomach) and non-cardia (lower stomach) subtypes.These entities differ in terms of risk factors, carcinogenesis and epidemiologic patterns.ChronicH.pyloriinfection is considered the principal cause of non-cardia gastric cancer, being nearly all cases attributed to this bacterium[48,49].The worldwide prevalence ofH.pyloriinfection is extraordinarily high, affecting around 50% of population[50], and its geographic variability correlates with the incidence of gastric cancer.Yet, less than 5% of infected persons will develop cancer, likely because of differences in bacterial and host genetics, age of infection and environmental factors[51].In 1994, the World Health Organization categorizedH.pylorias a Carcinogen type I for gastric cancer on the basis of observational studies reported from the International Agency for Research in Cancer[52].
H.pyloriinfection has been associated with an increased risk of all variants of adenocarcinoma, whether diffuse or intestinal, and whether located in the body of the stomach or in the antrum; actually, it is related to tumors distal to the cardia.Conversely, tumors growing in the esophagogastric junction, usually arising from the altered mucosa in Barrett’s esophagus, have not been linked toH.pyloriinfection[53].
H.pylorisurvives in the gastric acid microenvironment, being able to damage the mucosa.It induces a chronic inflammatory response that results in chronic gastritis and peptic ulceration.Development of gastric cancer is one of the long-term consequences ofH.pyloriinfection[54].ChronicH.pyloriinfection has been also related to gastric MALT (Mucosa associated Lymphoma), which is an extra-node lymphoma variant consisting of a morphologically heterogenous small B cells neoplasm[55].
H.pyloriis able to dysregulate host signaling pathways and to promote oncogenesis by two main mechanisms: Citotoxin-associated gene A (CagA) and its pathogenic island (Cag PAI); and vacuolating cytotoxin A (VacA).CagAH.pylori-protein is encoded by one of the genes located in the CagPAI island.CagA binds to the ectodomain of alfa5beta1 integrin allowing its internalization.Once it has been translocated, CagA binds to the inner surface of the cell membrane and undergoes tyrosine phosphorylation.Both the phosphorylated CagA and the unphosphorylated CagA can interact with a number of host proteins to activate downstream signaling pathways such as the RAS/MEK/ERK pathway, the NF-kB pathway, and the betacatenin pathway.Such pathways activation enhances the proliferation of gastric epithelial cells[56,57].
The VacA is secreted byH.pyloriand has a variety of biological functions.It binds to host cells and is internalized creating vacuoles (large vesicles) inside the host cells.Thisvacuolizationprocess leads to the activation of endosomes and early lysosomes.Additionally, VacA can also be transferred to the mitochondria where it alters the membrane barrier that releases cytochrome c which finally activates the pro-apoptotic factor Bcl-2 associated X protein (Bax) leading to apoptosis.Therefore, the alteration of the membrane impermeabilization ends up in preventing apoptosis of gastric cells[58].Inside the mitochondria, VacA can also activate the dynamic related protein 1 which inhibits the activation of Bax mitochondria outer membrane impermeabilization that prevents death by apoptosis[59].Further, it can also disrupt the gastric epithelial cell-to-cell unions, which prevents T lymphocyte activation and proliferation in the lamina propria, and ultimately induces inflammation and carcinogenesis by disrupting autophagy in gastric cells[60].
In addition,H.pyloriinduces a chronic inflammatory state by upregulating many pro-inflammatory cytokines, like IL-1, IL-6, IL-8, TNF-alfa, and NF-kB[61].Among them, activation of NF-kB and upregulation of IL-8 are crucial[62].Suppressor gene P53 also plays an important role in gastric inflammation and carcinogenesis.Mutations of p53 have been related to the development of gastric cancer.In fact, inactivation of p53 gene is present in 40% of gastric tumors and has been specifically found in patients infected with the CagA positive strain ofH.pylori[63].H.pyloriinfection has also been related to epigenetic modification during gastric carcinogenesis by promoting hypermethylation of O6-mehylguanine DNA methyltransferase (MGMT).MGMT protein is essential for the repair of O6-methylguanine, which prevents mutations during DNA replication.Therefore, reduced levels of MGMT can increase mutagenesis in the gastric epithelium[64].Finally,H.pylorihas been linked to the oxidative stress that promotes DNA damage in gastric tissue by stimulating the production of intracellular reactive oxygen and nitrogen species in gastric tissues, which can damage DNA tumor suppressor genes like p53 and contribute to gastric carcinogenesis[65].
In summary, theH.pyloriinfection is one of the leading factors in the development of gastric carcinoma.Chronic inflammation induced byH.pyloriis related to cell proliferation, apoptosis, epigenetic changes of tumor suppressor genes, and alterations of the oxidative stress mechanisms.Eradication ofH.pylori, especially in endemic areas, should be a healthcare priority in order to decrease gastric cancer-related casualties.
GALLBLADER CANCER
Gallbladder cancer (GC) is a common hepatobiliary malignancy that carries very poor prognosis.It is the fifth commonest gastrointestinal tract cancer and is endemic is several countries, with the highest incidence rate reported in Delhi (India), Pakistan and Quito (Ecuador).It shows female preponderance[66], and genetic, infectious, and lifestyle factors have been associated with GC[67].Infection leads to chronic gallbladder inflammation that seems to contribute to carcinogenesis.Two species- Salmonella Typhi and Helicobacter Pylori- and a liver parasite -Clonorchis sinensis-, among others, have been specifically linked to GC.
Salmonella Typhi
The association between chronic typhoid infection and GC was first reported in 1971[68] and confirmed by ulterior studies.For example, in 1964, during the typhoid outbreak in Aberdeen (Scotland), Caygillet alstudied the mortality rate of infected population and reported a 6% of risk of death attributable to GC among chronic carriers[69].
Further studies from Northern India reported a 7.9-fold increased risk of GC among infected persons.Species of Salmonella typhi and paratyphi-A could be isolated from bile, gallbladder tissue and stones in patients with GC.Bacterial serologic detection was related to a 9.2-fold increased risk of developing GC.
Another study from India, using nested PCR technique in hepatobiliary specimens, detected a 67.3% of typhoid carriers among GC patients compared to 8.3% of controls.The authors concluded that the liver served as a preferential survival niche forSalmonellaTyphi (S.Typhi)[70].AsSalmonellaspp.are excreted in the gallbladder, the multiplying bacteria secrete toxins and metabolites that favor mutational changes.Several carcinogens have been proposed: bacterial glucuronidase, bacterial enzymes and the production of nitrous compounds from nitrates[71,72].
Additionally,S.Typhi produces a genotoxin, the cytolethal distending toxin (CDT) that allows bacteria to internalize inside the host cells.Then, they are able to avoid the usual endocytic pathway that leads to the formation of destructive lysosomes and finally reaches a specific intracellular compartment where it can survive and replicate[73].Once in the cytosol, the CDT facilitates the persistence of infection thanks to its immunomodulatory activity.Later on, it can reach the nucleus causing irreversible DNA damage[74].
Helicobacter spp.
H.bilis,H.pullorum,H.hepaticus,andH.pylorihave been suspected to cause biliary tract diseases.Helicobacterspp.cause persistent infection in the biliary tract inducing chronic inflammation and gallstone formation, mainlyviaurease production[75,76].Helicobacterspp.can produce several carcinogenic toxins and metabolites that initiate malignant cell transformation within the gallbladder[77].Helicobacter bilis specific DNA sequences have been amplified in 27.2% of gallbladder and in 33.3% of biliary duct cancers[77].
Escherichia coli
Escherichia coli(E.coli) is part of the regular human intestinal microbiota but may become highly pathogenic following the acquisition of virulence factors, like the cytotoxic necrotizing factor I -CDT I-.The presence of genotoxin colibactin, related toE.coliinfection, is known to be capable of inducing double stranded DNA breaks[10].
Opisthorchis viverrini and Clonorchis sinensis
Liver flukeClonorchis sinensis(C.sinensis) is a high-risk pathogenic parasitic helminth endemic in some Asian countries.This parasite is classified among the Group I of human carcinogens by the IARCC[1].Fresh water snails and several species of fish serve as secondary intermediate hosts.Humans and other fish-eating animals are infected through the ingestion of raw or undercooked freshwater fish that contains meta-cercariae.After ingestion, the meta-cercaria excysts in the duodenum and ascends the biliary tract through the ampulla of Vater[78].
Infection withOpisthorchis viverrinihas been related to cholangiocarcinoma upon the results of several cross-sectional and case-control studies[79].The association betweenC.sinensisinfection and cholangiocarcinoma is even better substantiated and the IARCC has already classified it as a probable carcinogenic for humans (Group 2A)[80,81].
Liver fluke pathogens cause mechanical injury and inflammation in the biliary tree, leading to metaplasia of mucin-producing cells, periductal fibrosis and hyperplasia of epithelial cells[82].The severity of these changes correlates with the duration of the infection, the parasite load and the susceptibility of the host.The molecular mechanisms involved in the development of cholangiocarcinoma are poorly known, and a multistep process has been proposed: chronic inflammation damages cholangiocytes which in turn promotes cell proliferation and genetic and epigenetic mutations, finally transforming cholangiocytes into malignant cells[83].Other carcinogenetic mechanisms include genomic instability, transcriptomic, proteomic and microRNA profile alterations and dysregulation of immune response.
LIVER CANCER
Worldwide, liver cancer is the sixth most common cancer, yet the second leading cause of cancer-related death in men, with 745000 deaths per year[84].Hepatocellular carcinoma (HCC) represents approximately 90% of all primary liver cancers.It preferentially affects males and presents a wide geographical variation.However, chronic Hepatitis B virus and HCV infections are the leading cause of HCC, being responsible for 60%-80% of all these tumors worldwide, especially in developing countries.
Hepatitis B virus (HBV) is a partially double-stranded DNA virus that replicatesviareverse transcription.It is highly contagious and is transmitted by percutaneous and permucosal exposure to infected blood and other body fluids.HBV particles package the incomplete double-stranded DNA into the nucleus of the host, where the virus is recognized as damaged and induces a DNA repair response, resulting in virus replication[85].Chronic HBV infection is one of the leading causes of HCC worldwide.HBV typically replicates inside the hepatocytes.Virions bind to the surface of the hepatocytes and the nucleocapsid is released into the cytoplasm and translocated by microtubules to the microtubule-organizing center near the nucleus.Once inside the nucleus, HBV can lead to histone degradation, which enhances chromatin dynamics and may promote genetic instability, chromosomal alterations, and can also initiate oncogene mutation[86].However, HBV integration into the hepatocytes occurs randomly -at one or multiple sites- and occasionally may promote direct oncogene activation or inactivate tumor suppressor genes[87].While viral integration is an early event, selective clonal amplification of hepatocytes occurs throughout progression of the disease[88].It is known that both the HBV-encoded envelope and the regulatory HBx protein directly contribute to hepatocyte transformation: HBx regulates expression of many genes involved in signal transduction pathways, cell cycle control, metastases, and avoidance of immune response.Several cohorts and case-control studies have assessed the risk of HCC among individuals infected with HBV[89,90].The ability of antivirals to inhibit HCC progression is limited, likely because hepatocarcinogenesis seems to occur prior to the onset of liver fibrosis/cirrhosis.Therefore, once the disease is established, there is no turning back.
HCV is a single-stranded, positive sense RNA virus that encodes a single polyprotein that can form structural -which constitute the viral particle, such as the core protein- and non-structural proteins which support viral genomic replication.Seven genotypes have been described.Epidemiological studies show than infection with genotypes 1b and 3 is associated with an increased risk of developing HCC[91].HCV replicates in the cytoplasm of hepatocytes and is unique among cancer-causing viruses by not encoding oncoproteins or integrating its genome into the host chromosomal DNA.In fact, HCV core genes variants have been associated with HCC even in patients in which the infection had already disappeared[92].This suggests that viral factors influence progressive liver disease.HCV associated carcinogenesis includes increased hepatocyte proliferation and steatosis; virus-induced inflammation and oxidative stress which induces genomic mutations and genome instability; mitochondrial damage and induction of reactive oxygen species; and inhibition of host immune responses[93].HCC is associated with the development of multifocal, genetically distinct tumors throughout the liver, suggesting that the entire organ is altered.
COLON CANCER
Colorectal cancer is the leading cause of mortality regarding gastrointestinal neoplasms worldwide[94].Pathogenic microorganisms able to induce intestinal dysbiosis have become the spotlight of current research in this area, as they carry the potential for colorectal tumorigenesis.Fusobacterium nucleatum,E.coli,Bacterioides fragilis(B.fragilis) andSalmonella enterica(S.enterica) have been reported as high-risk oncogenic pathogens.
Fusobacterium nucleatum
Fusobacterium nucleatum(F.nucleatum) is an adherent and invasive Gram-negative anaerobic bacterium frequently found in the oral cavity.Current research relatesF.nucleatumto the development and progression of colon cancer, and has been found in primary lesions and stools of patients with colon cancer[95], especially those located in the cecum and rectum[96].Tumoral cells over-express Gal-GalNAc molecules, which promote bacterial adhesion through the Fap2 protein[97].
Additionally,F.nucleatuminfection has been related to a decreased survival rate among colon cancer patients and an increased resistance to chemotherapy agents[98,99].The presence ofF.nucleatumin colon cancer patients varies worldwide, ranging from 15% in North American to 60% in Chinese patients[100].Interestingly, tumors infected withF.nucleatumshow three similarities: they are microsatellite unstable, show a methylation phenotype of CpG island, and exhibit mutations in the BRAF/KRAS genes[101].Microsatellite instability is responsible for the ability of infected cells to elude the immune response[102] and has also been related to the activation of beta-catenin signaling pathway, commonly unregulated in colon cancer[103].F.nucleatumalso promotes inflammation, by increasing TNF-alfa and IL-10 Levels in adenomas and Il-6 and IL-8 in carcinomas, both regulated by the NF-kB transcription factor[104].
E.coli
E.coliis a widely distributed Gram-negative bacterium that can alter the intestinal microbiome.The B2E.colistrains have been related to colon cancer[105].E.colipromotes colon pathologic inflammation -as in Chron’s disease- which seems to be a relevant factor in colon cancer formation[106].However, the exact role ofE.coliin the pathogenesis of colon cancer is not completely known.Recent studies have identified two potentially pathogenicE.colistrains: Adherent-invasiveE.coli(AIEC) and enteropathogenicE.coli(EPEC).During infection, AIEC binds to CEACAM6 (a cellular adhesion receptor associated to carcinoembryonic antigen- CEA- which is overexpressed in colon cancer cells[107].Infection stimulates IL6 production, which together with the increased expression of CEACAM6 can promote carcinogenesis.AIEC also secretes colibactin, a secondary metabolite associated to DNA damage acting as an alkylating agent and promoting tumor development[108].EPEC is thought to stimulate macrophage-inhibitory cytokine-1 production - a cytokine related to metastasis- and inducing autophosphorylation of EGFR receptor[109,110].
B.fragilis
TheBacteroidesspp.are regularly found in the human intestine and comprises 30% of the microbiota[111].B.fragilisis an anaerobic Gram-negative bacterium colonizing about 0.5%-2% of the entire human intestine.A toxigenicB.fragilisstrain -also known as ETBF- has a pathogenicity island encoding a metalloproteinase, theB.fragilistoxin (BFT) which is associated to increased inflammatory bowel disease and colitis, both considered high risk factors to develop colon cancer[112].B.fragilishas also been detected in stool from cancer patients.Although the exact role of enterotoxigenicB.fragilishas not been completely described, it is believed that carcinogenesis is either induced by BFT toxin secretion or through host immune system dysregulation[113].It can also trigger carcinogenesis through the beta catenin pathway activation by disrupting the adherent e-cadherin gap unions[114].
S.enterica
S.entericaincludes serotypes of Salmonella typhi, Salmonella paratyphi, Salmonella enteritidis and Salmonella typhimurium.In recent years, it has been found that bacteria may modulate host immune response in two different ways: First, it can promote carcinogenesis inducing both DNA damage and increasing cell abnormal proliferation; and second, it can induce cell migration as a result of chronic inflammation[17].
Two proteins ofS.entericahave been associated with an increased risk of developing colon cancer: typhoid toxin and AvrA effector protein.Typhoid toxin is a cyclomodulin that, similar toE.coliCDT, increases cell survival and promotes intestinal dysbiosis, resulting in the development of inflammatory bowel disease and colon cancer[115,116].AvrA is secreted by bacteria and has been found in stool samples from colon cancer[117].AvrA may promote inflammatory and immune dysregulation through several mechanisms: the inhibition of NF kappa-beta signaling pathway, the inhibition of IL-12, INF-c and TNF-α secretion, the inhibition of IL-6 transcription, and the increase of IL-10 transcription[118,119].On the other hand, AvrA might also activate the Wnt/β catenin pathway inducing cellular proliferation, by both β catenin phosphorylation and deubiquitination[120].
ANAL CANAL
Anal cancer refers to the malignancy of the intestinal mucosa arising in the anatomic anal canal, defined as beginning at the dentate line and ending at the anal verge.Eighty-five percent of anal tumors are of squamous cell origin, approximately 10% are adenocarcinomas, and the remaining 5% are other neoplasms (melanoma, small cell carcinoma…)[121].Although it comprises 2.7% of all gastrointestinal malignancies, anal cancer incidence has been increasing over the last few decades[122].
As previously highlighted in the paper, HPV are small DNA viruses that infect various epithelial tissues.They also include the anogenital tract, and HPV is recognized as the causative agent of more than 80% of cases of anal cancer[123].The difference in their ability to promote malignant transformation is the basis for the classification of HPV into low and high-risk variants.Mainly two HPV oncogenic subtypes, 16 and 18, are related to the development of squamous anal cancer[124].HPV can integrate into the host DNA.Epithelial cells that harbor integrated HPV 16 DNA have a selective growth advantage over cells that carry normal extrachromosomal viral genomes; this growth advantage correlates with the increased expression of two viral genes in particular, E6 and E7[125].The early proteins, E6 and E7, bind and inactivate the tumor-suppressor gene p53, and the retinoblastoma tumorsuppressor protein (pRb), respectively[126-128].
GUT MICROBIOTA
Most of the literature summarized in the present paper has been published previous to 2016 as research over the last years is focused in the role of gut microbiota in human carcinogenesis.Although it is not the scope of our review, we cannot conclude our review without summarizing intestinal microbiota key points.Gut microbiota consists of viruses, fungi and more than 1000 bacteria which are crucial for maintaining the gut barrier, metabolism and immunity.Disruption of the host relationship with the microbiota might result in oxidative stress, chronic inflammation and dysbiosis which can finally promote carcinogenesis[129].The influence of gut microbiota relies mainly on cancers in the gastrointestinal tract, and the regulation of microbiota by diet, prebiotics, probiotics, symbiotics and antibiotics are proposed as new strategies to be explored in the future[130].Gut microbiota is thought to play an important role in colon -stool samples of patients with CRC have higher proportions of Escherichia and Fusobacterium and lower concentrations of Firmicutes and Actinobacteria[131], HCC[132] and GC[133], Interactions between gut microbiota and GI cancers are likely to yield new opportunities to reduce cancer morbidity and mortality (Table 1).
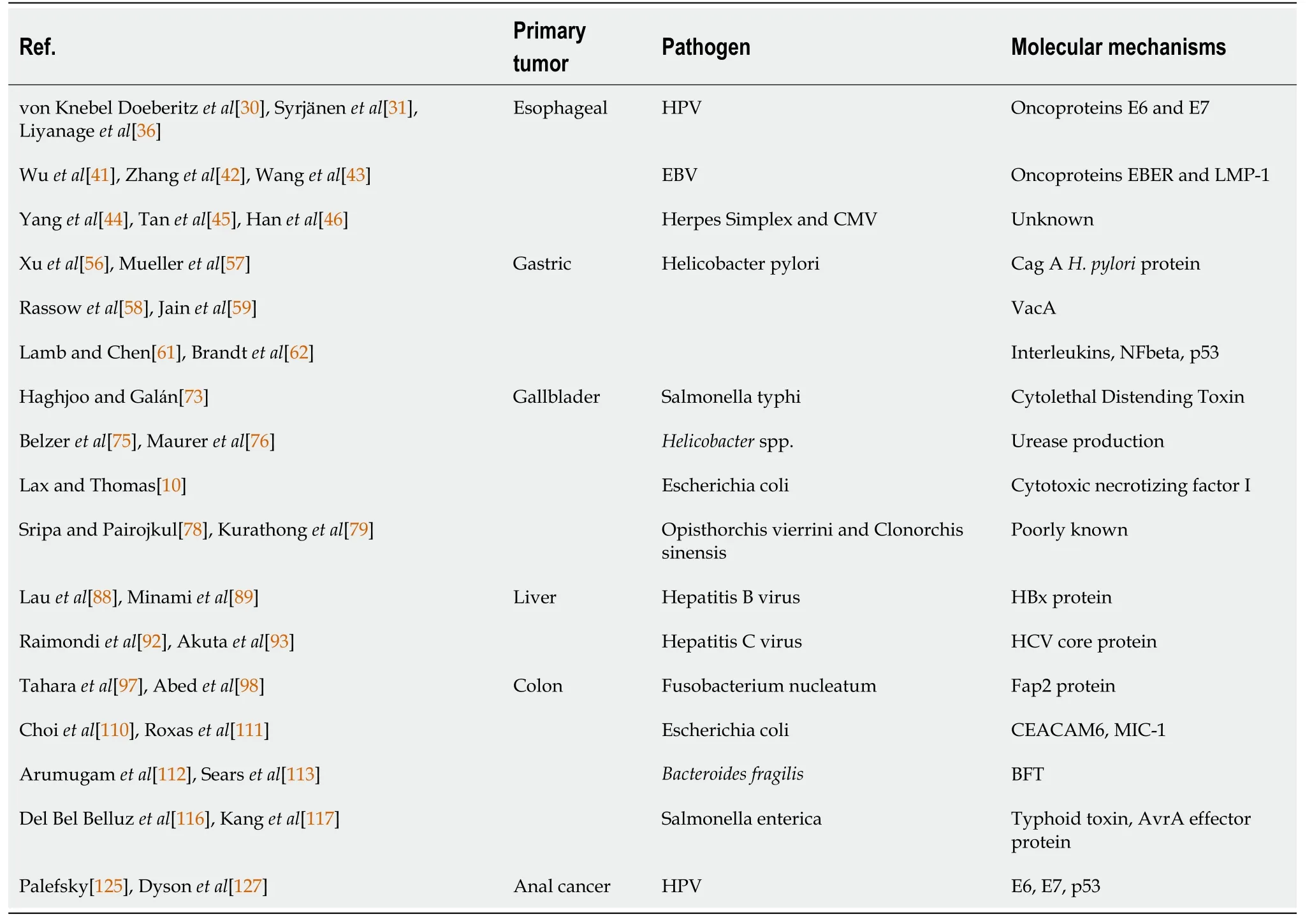
Table 1 Pathogens related to gastrointestinal malignancies
CONCLUSION
Cancers arise from the transformation of a single cell so that its behavior is no longer under the control of normal regulatory pathways.The relationship between some pathogens and gastrointestinal tumor carcinogenesis has been well stablished.However, this relation is difficult to determine with many other agents as cancer is a multistep process.Besides, the presence of bacteria at the site of a tumor does not itself implies causation.There is a long time period between the onset of carcinogenesis and the development of overt disease, and randomized studies are expensive and extremely difficult to make (one cannot, for example, infect a person with an agent and then wait to see if cancer develops).However, the occurrence of cancer attributable to infections is a global concern especially in underdeveloped regions.Chronic infections can mimic precancerous lesions that could be treated with antibiotics or antiviral treatment and therefore prevent the onset of carcinogenesis.Greater understanding of the consequences of long-term infections will help to elucidate the exact pathogenic processes involved in the development of some pathogen-related neoplasms.Preventive measures (like vaccines and antimicrobial agents) aimed to eradicate these oncogenic microorganisms should ideally interrupt the carcinogenetic pathways and avoid the formation of a relevant proportion of gastrointestinal cancers.Only global effort and an international strategy might help to decrease gastrointestinal cancer related deaths secondary to infectious pathogens.New molecular techniques are needed to identify new infectious agents in tissues.However, caution is requested not to overemphasize the association between pathogens and gastrointestinal malignancies without a proper causation proof.
 World Journal of Meta-Analysis2021年6期
World Journal of Meta-Analysis2021年6期
- World Journal of Meta-Analysis的其它文章
- Simulating the mind and applications - a theory-based chance for understanding psychic transformations in somatic symptom disorders
- Is dose modification or discontinuation of nilotinib necessary in nilotinib-induced hyperbilirubinemia?
- Preclinical safety, effectiveness evaluation, and screening of functional bacteria for fecal microbiota transplantation based on germ-free animals
- Newer developments in viral hepatitis: Looking beyond hepatotropic viruses
- Prediabetes and cardiovascular complications study: Highlights on gestational diabetes, self-management and primary health care
- Hydroxychloroquine alone or in combination with azithromycin and corrected QT prolongation in COVID-19 patients: A systematic review