
乳腺浸润性导管癌潜在核心基因的生物信息学分析
2021-02-01 02:50罗茂珍
实用癌症杂志 2021年1期
罗茂珍 权 毅
乳腺浸润性导管癌(IDC)是乳腺癌主要亚型,发病率逐年上升,且呈现年轻化趋势[1-2]。目前对于IDC发生、转移具体机制不详。全基因组分子图谱能够揭示肿瘤发生发展过程中的分子变化,并已被证明是识别关键基因的一种高效方法。本研究旨在通过生物信息学方法对从公共基因芯片数据库(GSE29044)下载的IDC组织与正常乳腺组织的芯片数据进行分析,探讨在IDC发生和预后中潜在的关键基因和关键途径。
1 资料与方法
1.1 资料
本研究从美国国立生物技术信息中心(NCBI)的公共基因芯片数据库(GEO)[3]下载编号为GSE79058基因芯片数据,该芯片包含了103个样本,其中67个IDC样本数据,36个正常乳腺组织样本数据,并均由GPL570实验平台(Affy-metrix公司的HG-U133-Plus-2芯片)进行分析。
1.2 方法
1.2.1 差异基因分析 用GEO2R在线分析工具分析IDC与正常乳腺组织样本的差异基因,并计算P值及logFC值。设置P<0.01、logFC>2或<-2获得DEGs。
1.2.2 GO功能及KEGG通路富集分析 GO分析大规模功能是富集研究中常用的一种方法,基因功能可分为细胞成分(cellular component,CC)、分子功能(molecular function,MF)和生物过程(biological process,BP)。KEGG是一个广泛应用的数据库,它存储了大量有关基因组、生物途径、疾病、化学物质和药物的数据。应用DAVID在线数据库[4]的GO功能、KEGG功能对DEGs进行分析。
1.2.3 构建差异表达基因的蛋白质-蛋白质相互作用网络(PPI) 应用STRING在线数据库[5]对DEGs进行PPI分析,将结果导入Cytoscape软件可视化。运用Cytoscape软件中的MCODE插件构建模型,筛选出连接度较高的枢纽基因。
1.2.4 枢纽基因的生存分析 运用Kaplan-Meier在线绘图仪生存分析工具来评估枢纽基因在乳腺癌患者中的预后价值。根据mRNA表达的中位数,将癌症患者分为2组。P<0.05为差异有统计学意义。
2 结果
2.1 DEGs筛选
根据P<0.01,logFC>2或<-2筛选出398个DEGs,其中有110个上调差异基因和288个下调差异基因(火山图见图1)。

图1 差异基因火山图
2.2 DEGs的GO功能和KEGG富集分析
使用DAVID在线数据库对DEGs进行GO功能和KEGG富集分析。在生物过程方面,上调差异基因主要富集在核有丝分裂、细胞分裂、细胞增殖、胶原蛋白分解代谢的过程、内皮细胞分化、有丝分裂细胞周期的G2/M转变和促进后期复杂依赖的分解代谢过程,下调差异基因富集在细胞趋化作用、细胞粘附、积极的调控细胞增殖和半桥粒装配。细胞组分方面,上调差异基因富集在细胞中间体、纺锤体、纺锤体微管和蛋白质的细胞外基质,下调差异基因富集在细胞外空间、细胞外区域、细胞外基质外来体、蛋白质的细胞外基质和受体复合物。在分子功能方面,上调差异基因富集在蛋白结合、ATP结合和蛋白丝氨酸/苏氨酸激酶活性,下调差异基因主要是与在肝素结合。上调差异基因富集在细胞周期、卵母细胞减数分裂、细胞外基质受体相关作用和p53信号通路,下调差异基因富集在细胞黏着斑连接联和PPAR信号通路(结果见表1、2,图2、3所示)。

图2 差异基因GO功能富集结果
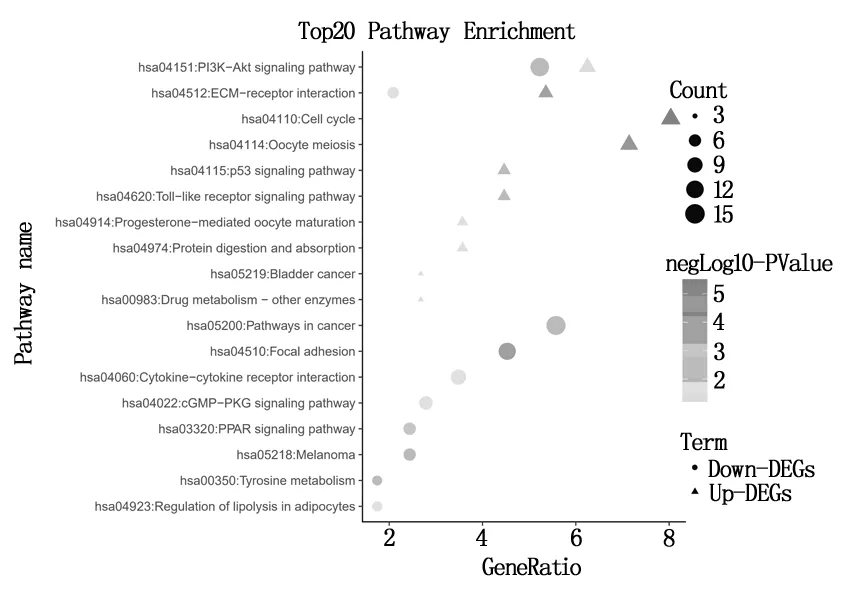
图3 差异基因信号通路富集结果

表1 下调差异基因富集分析

表2 上调差异基因富集分析
2.3 蛋白互作网络(PPI)分析和枢纽基因筛选
使用STRING在线数据库分析DEGs,构建复杂DEGs相互作用网络,PPI网络中共有348个节点和2090条边,将结果导入Cytocscape(结果见图4)。为了在PPI网络中检测出重要的聚类模块,使用MCODE插件进行模块分析,得分最高的模块1中包含42个蛋白(结果见图5),均为上调基因编码。从模块1中筛选出连接度较高的HMMR、CDK1、PBK、CCNB2、TPX2、AURKA、DLGAP5、NUSAP1、TOP2A和CEP55等10个与乳腺癌研究较少的枢纽基因。
2.4 枢纽基因生存分析
将HMMR、CDK1、PBK、CCNB2、TPX2、AURKA、DLGAP5、NUSAP1、TOP2A和CEP55等10个枢纽基因上传至Kaplan-Meier在线绘图仪生存分析平台,HMMR、CDK1、PBK、CCNB2、TPX2、AURKA、DLGAP5、NUSAP1、TOP2A和CEP55的高表达均与乳腺癌不利总体生存相关(结果见图6)。

图4 差异基因蛋白互作网络

图5 模块1的42个基因(得分40.39,42个节点,828条线)
3 讨论
本研究通过从GEO数据库里获取乳腺浸润性导管癌(IDC)组织与正常乳腺组织的基因表达芯片数据,对二者的差异基因(DEGs)进行分析,筛选出得分最高的模块1,主要参与核有丝分裂及调控、纺锤体、减数分裂细胞周期过程、细胞中间体、促进后期复杂依赖的分解代谢过程和微管细胞骨架组织参与有丝分裂等生物过程。参与细胞周期、卵母细胞减数分裂和p53信号通路等信号通路。最终筛选出HMMR、CDK1、PBK、CCNB2、TPX2、AURKA、DLGAP5、NUSAP1、TOP2A和CEP55等10个高表达与乳腺癌不利总体生存相关的枢纽基因。透明质酸介导的运动受体(HMMR)是一种以透明质酸(HA)为底物与微管蛋白相互作用的多功能细胞外蛋白,HMMR具有调节细胞运动,干扰细胞有丝分裂,促进血管生成等作用[6],其表达水平与结膀胱癌[7]、胃癌[8]等癌症的潜在不良预后有关。同时HMMR具有抗肿瘤作用,为结直肠癌等癌症提供抗原表位,有望成为免疫治疗的靶点[9]。而HMMR与乳腺癌关系的研究目前较少,Liu等[10]的研究发现HMMR-AS可以抑制Basal-like型乳腺癌细胞的增殖和转移,有可能成为其潜在治疗靶点。Liu等[11]研究发现抑制细胞周期蛋白依赖性激酶1(CDK1)能够抑制三阴性乳腺癌的肿瘤生长和诱导凋亡。Tonnessen-Murray C[12]、Li Jie[13]认为CDK1、人细胞周期蛋白B2(CCNB2)可能通过调节G2/M转换参与乳腺癌的发生过程。CCNB2[13]、CDK1[14]的高表达与乳腺癌恶性程度成正相关。研究认为[15]在乳腺癌中丝氨酸/苏氨酸激酶Akt(PKB)通过PI3K/PKB信号通路调节癌细胞浸润和转移,通过抑制此通路可诱导细胞凋亡,因此成为近年来乳腺癌分子靶向治疗研究热点。TPX2参与细胞有丝分裂纺锤体微管形成过程,Yang等[16-17]的研究发现TPX2在乳腺癌中明显高表达,促进乳腺癌细胞增殖、迁移和侵袭。高水平TPX2与三阴性乳腺治疗后总生存期(OS)相关[18]。有丝分裂极光激酶(AURKA)促进G2/M事件,且AURKA通过增强乳腺癌干细胞(BCSC)表型,在恶性细胞中引起明显的致癌特性[19]。核仁和纺锤体相关蛋白1(NUSAP1)可通过调节AURKA影响乳腺癌细胞的增殖、转移,下调NUSAP1可增强乳腺癌对表阿霉素的敏感性[20]。XU[21]、Yang[22]认为乳腺癌患者TOP2A的低表达较高表达有更好的无病生存期(DFS)和总生存期(OS)。在HER-2阳性乳腺癌患者中,TOP2A若扩增,采用蒽环类药物进行新辅助化疗可能改善预后[23]。中心体相关蛋白(CEP55)是细胞有丝分裂的关键调控因子,其过表达与基因组不稳定相关[24]。在小鼠实验中,MiR-144通过抑制CEP55抑制乳腺癌细胞的侵袭和转移[25]。目前CEP55与乳腺癌相关临床研究较少,在本研究中CEP55在IDC中高表达,且与乳腺癌OS不良结果相关。

图6 HMMR、CDK1、PBK、CCNB2、TPX2、AURKA、DLGAP5、NUSAP1、TOP2A和CEP55表达与乳腺癌患者生存的关系
综上所述,通过GEO芯片数据库进行基因差异分析,我们发现HMMR、CDK1、PBK、CCNB2、TPX2、AURKA、DLGAP5、NUSAP1、TOP2A和CEP55等10个枢纽基因,枢纽基因过表达均与乳腺癌患者总体生存差相关。因此,通过差异分析有助于发现乳腺癌潜在发病基因和发病机制,这些枢纽基因可能成为乳腺癌的预后生物标志物和潜在治疗靶点,但仍有待于大量前瞻性临床研究和实验验证。
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2021年10期)2021-12-02
健康之家(2021年19期)2021-05-23
房地产导刊(2020年8期)2020-09-11
心电与循环(2020年1期)2020-02-27
商周刊(2019年18期)2019-10-12
家庭影院技术(2019年4期)2019-04-17
中国生殖健康(2019年5期)2019-01-06
当代陕西(2018年12期)2018-08-04
江苏农业科学(2017年5期)2017-04-15
