
HPLC-QAMS法同时测定前列平胶囊中7种成分的含量
2021-01-27 01:56刘德军樊苗苗陶弘武慕扬娜
实用药物与临床 2020年12期
刘德军,贺 玲,樊苗苗,陶弘武,慕扬娜
0 引言
前列平胶囊主要由北败酱、红花、丹参、赤芍、桃仁、泽兰、石韦、乳香和没药9味中药组方而成,临床上主要用于湿热瘀阻所致的急、慢性前列腺炎的治疗[1-4]。前列平胶囊方中北败酱清热解毒、祛瘀止痛,为君药;红花、桃仁活血祛瘀、润肠止痛,赤芍清热凉血,为其臣药;丹参活血祛瘀、凉血消痈,乳香、没药散瘀定痛、消肿生肌,泽兰祛瘀消痈、利水消肿,合为佐药;石韦利尿通淋、凉血止血,引诸药直达病所,为使药,诸药合奏,以达清热利湿、化瘀止痛的临床功效。前列平胶囊现行质量标准和文献报道[5]中仅对芍药苷、丹酚酸B进行了定量研究,未对方中君药北败酱及其他药味所含成分进行研究。中药及其制剂具有多组分、多靶点、整体协同作用的特点,近年来,多指标成分质量评价模式已广泛应用于其质量控制中。本研究按照中药质量标志物确认原则,采用HPLC-QAMS法对前列平胶囊方中君药北败酱所含代表性成分菊苣酸,臣药红花所含特征成分羟基红花黄色素A和红花黄色素A,佐药丹参所含主要活性成分丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA含量进行同时测定,为全面评价前列平胶囊质量提供科学依据。
1 材料
Agilent 1200型高效液相色谱仪(美国安捷伦公司)、Shimadzu LC-20A型高效液相色谱仪(日本岛津公司);Agilent ZORBAX SB-C18色谱柱(250 mm×4.6 mm,5 μm)、Phenomenex C18色谱柱(250 mm×4.6 mm,5 μm)、Eclipse XDB-C18色谱柱(250 mm×4.6 mm,5 μm);XS105DU型电子分析天平(瑞士梅特勒-托利多公司);KQ-100DE型数控超声波清洗器(昆山市超声仪器有限公司)。菊苣酸对照品(批号:111752-201703,CAS号:6537-80-0,含量:97.6%)、羟基红花黄色素A对照品(批号:111637-201810,CAS号:78281-02-4,含量:93.1%)、丹酚酸B对照品(批号:111562-201917,CAS号:121521-90-2,含量:96.6%)、丹参素钠对照品(批号:110855-201915,CAS号:67920-52-9,含量:97.8%)和丹参酮ⅡA对照品(批号:110766-201721,CAS号:568-72-9,含量:99.5%)均来源于中国食品药品检定研究院;丹参酮Ⅰ对照品(批号:17051706,CAS号:568-73-0,含量:99.9%)来源于上海同田生物技术股份有限公司;红花黄色素A对照品(批号:CFS201801,CAS号:85532-77-0,含量:98.5%)来源于武汉天植生物技术有限公司;乙腈为色谱纯,其余试剂均为分析纯;前列平胶囊[规格:每粒装0.42 g(相当于饮片2.2 g),批号:20190207、20190402、20190403]来源于西安千禾药业有限责任公司。
2 方法与结果
2.1 单组分对照品储备液和混合对照品溶液的制备 精密称取菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素钠、丹酚酸B、丹参酮 Ⅰ 和丹参酮 Ⅱ A对照品各适量,用70%甲醇溶液分别制成菊苣酸0.132 mg/ml、羟基红花黄色素A 0.918 mg/ml、红花黄色素A 0.774 mg/ml、丹参素0.162 mg/ml、丹酚酸B 2.526 mg/ml、丹参酮Ⅰ 0.218 mg/ml、丹参酮Ⅱ A 0.356 mg/ml的单组分对照品储备液;精密吸取单组分对照品储备液各2.5 ml,用70%甲醇溶液定容至50 ml,制成各成分质量浓度分别为6.6、45.9、38.7、8.1、126.3、10.9和17.8 μg/ml的混合对照品溶液。
2.2 供试品溶液和阴性样品溶液的制备 取前列平胶囊10粒内容物,研细混匀,精密称取0.5 g,置25 ml量瓶中,加70%甲醇20 ml,超声处理30 min,放冷后用70%甲醇定容至刻度,过滤制得前列平胶囊供试品溶液。按前列平胶囊质量标准WS-10459(ZD-0459)-2012Z-2018项下的工艺处方,分别制备不含北败酱、红花和丹参的3个阴性样品,再按上述方法制成阴性样品溶液。
2.3 色谱条件及专属性试验 色谱柱:Agilent ZORBAX SB-C18柱(250 mm×4.6 mm,5 μm),柱温为30 ℃;流动相:乙腈(A)-0.4%磷酸水溶液(B),梯度洗脱(0~11.0 min,14.0%A;11.0~15.0 min,14.0%A→22.0%A;15.0~24.0 min,22.0%A→30.0%A;24.0~45.0 min,30.0%A→72.0%A;45.0~50.0 min,72.0%A→14.0%A),流速为1.0 ml/min;检测波长分别为330 nm(0~15.0 min检测菊苣酸)[6]、403 nm(15.0~24.0 min检测羟基红花黄色素A和红花黄色素A)[7]和280 nm(24.0~50.0 min检测丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA)[8-10];进样量:10 μl。精密吸取“2.1~2.2”项下各溶液依法进样检测,结果主成分菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素钠、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA色谱峰峰形对称,理论塔板数按各成分色谱峰计均不低于4 000,主成分峰与相邻色谱峰均能有效分离,分离度均>1.5,阴性样品对前列平胶囊中7种成分的测定无干扰,色谱图见图1。
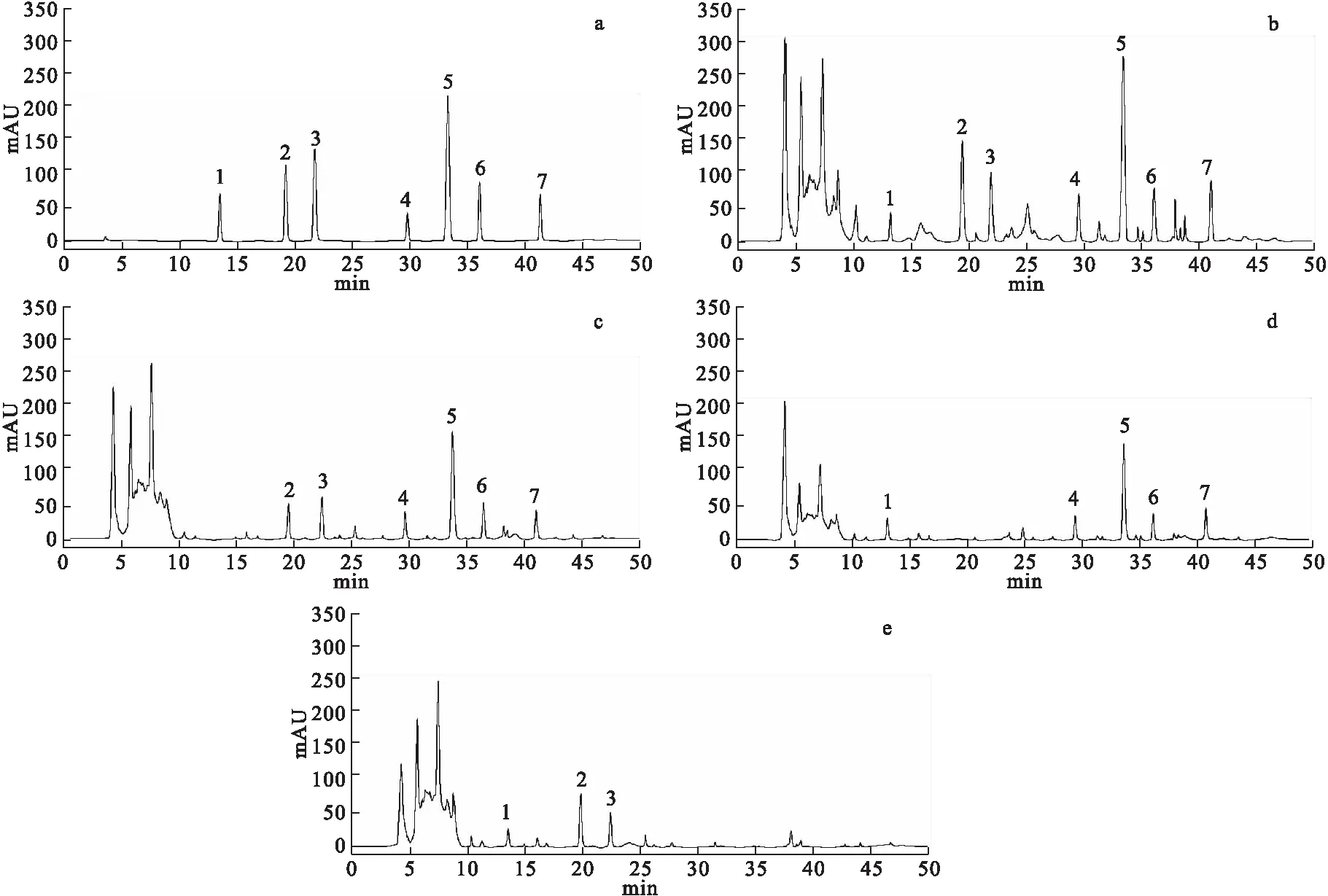
图1 专属性试验HPLC图注:a.混合对照品,b.前列平胶囊,c.北败酱阴性样品,d.红花阴性样品,e.丹参阴性样品。1.菊苣酸,2.羟基红花黄色素A, 3.红花黄色素A,4.丹参素,5.丹酚酸B,6.丹参酮 Ⅰ,7.丹参酮 ⅡA
2.4 线性关系考察 分别精密吸取“2.1”项下单组分对照品储备液各0.1、0.5、1.0、1.5、2.0、2.5 ml,置20 ml量瓶中,用70%甲醇溶液制成系列混合对照品溶液,依法进样测定菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮 ⅡA的峰面积,以质量浓度为横坐标,峰面积为纵坐标,进行线性回归,结果见表1。

表1 7个成分线性关系和范围
2.5 进样精密度、重复性及稳定性考察 取“2.1”项下混合对照品溶液重复进样6次,测得菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA色谱峰峰面积的RSD分别为1.35%、0.89%、1.01%、1.26%、0.58%、1.11%和0.97%。
取同一批号前列平胶囊样品,按“2.2”项下方法平行制备供试品溶液6份,依法进样测定菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA的峰面积,计算得7种成分含量的RSD分别为0.81%、1.34%、1.40%、1.72%、0.96%、1.58%和1.63%。
取前列平胶囊同一份供试品溶液,于制备后0、2、4、6、12、18 h、24 h进样检测菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA的峰面积,结果前列平胶囊供试品溶液24 h内稳定,7种成分峰面积的RSD分别为1.33%、0.91%、0.99%、1.25%、0.57%、1.08%和0.95%。
2.6 加样回收率试验 取已知含量的同一批次前列平胶囊内容物,研细混匀,取9份,每份0.25 g,精密称定,置25 ml量瓶中,按《中国药典》2015年版四部要求,分别按已知含有量的50%、100%、150% 3个水平精密加入加标对照品溶液(菊苣酸0.074 mg/ml、羟基红花黄色素A 0.626 mg/ml、红花黄色素A 0.422 mg/ml、丹参素0.114 mg/ml、丹酚酸B 1.818 mg/ml、丹参酮Ⅰ 0.146 mg/ml、丹参酮ⅡA 0.238 mg/ml)0.5、1.0、1.5 ml各3份,再按“2.2”项下方法制备加样品溶液。
在上述色谱条件下进行检测,结果所测成分菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA的平均加样回收率及RSD分别为97.86%(1.44%)、99.42%(0.96%)、99.18%(0.83%)、96.97%(1.11%)、100.02%(0.59%)、97.91%(1.37%)和98.90%(0.76%)。
2.7 相对校正因子的测定及耐用性考察
2.7.1 相对校正因子的测定 精密吸取“2.4”项下6个混合对照品溶液,依法进样检测菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA的峰面积,以丹酚酸B为内参物,按照相对校正因子计算公式:fk/s=fk/fs=(Xk×Ys)/(Xs×Yk)(式中X代表质量浓度,Y代表峰面积,k代表内参物,s代表其他成分)计算菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹参酮Ⅰ和丹参酮ⅡA的相对校正因子,结果见表2。

表2 各成分的相对校正因子
2.7.2 不同仪器、不同色谱柱对相对校正因子的影响 精密吸取“2.1”项下混合对照品溶液,依法进样检测菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA的峰面积,考察Shimadzu LC-20A型、Agilent 1200型高效液相色谱仪和Agilent ZORBAX SB-C18(250 mm×4.6 mm,5 μm)、Phenomenex C18(250 mm×4.6 mm,5 μm)、Eclipse XDB-C18(250 mm×4.6 mm,5 μm)色谱柱对相对校正因子的影响,结果见表3。

表3 不同仪器、不同色谱柱对相对校正因子的影响
2.7.3 不同柱温对相对校正因子的影响 精密吸取“2.1”项下混合对照品溶液,依法进样检测菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA的峰面积,考察不同柱温28 ℃、29 ℃、30 ℃、31 ℃、32 ℃对相对校正因子的影响,结果见表4。
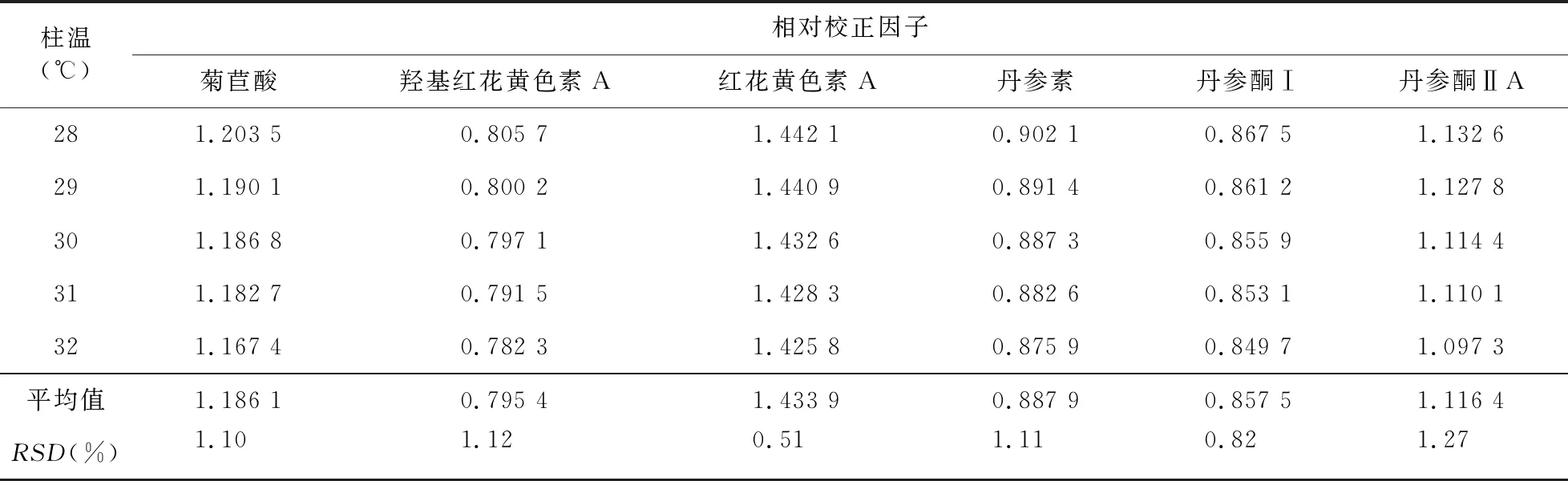
表4 不同柱温对相对校正因子的影响
2.7.4 不同体积流量对相对校正因子的影响 精密吸取“2.1”项下混合对照品溶液,依法进样检测菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA的峰面积,考察不同流速(0.8、0.9、1.0、1.1、1.2 ml/min)对相对校正因子的影响,结果见表5。

表5 不同体积流量对相对校正因子的影响
2.8 待测组分色谱峰的定位 精密吸取“2.1”项下混合对照品溶液,依法进样检测,记录菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA的保留时间,采用相对保留时间值法对待测成分色谱峰进行定位,考察Shimadzu LC-20A型、Agilent 1200型高效液相色谱仪和Agilent ZORBAX SB-C18(250 mm×4.6 mm,5 μm)、Phenomenex C18(250 mm×4.6 mm,5 μm)、Eclipse XDB-C18(250 mm×4.6 mm,5 μm)色谱柱对相对保留时间值的影响,结果见表6。

表6 不同仪器和不同色谱柱待测成分色谱峰的相对保留值
2.9 一测多评法与外标法测定结果比较 取3个批次的前列平胶囊样品,每个批次平行制备3份前列平胶囊供试品溶液,依法进样测定菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA的峰面积,首先采用外标法(ESM)计算7种成分的含量,再以丹酚酸B为内参物,按照“2.7.1”项下相对校正因子计算菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹参酮Ⅰ和丹参酮ⅡA的含量,结果见表7。结果显示一测多评法计算值与外标法实测值无明显差异(RAD<2.0%),表明所建立的相对校正因子可信度较高,所建立的一测多评法可用于前列平胶囊中菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA含量的同时测定。
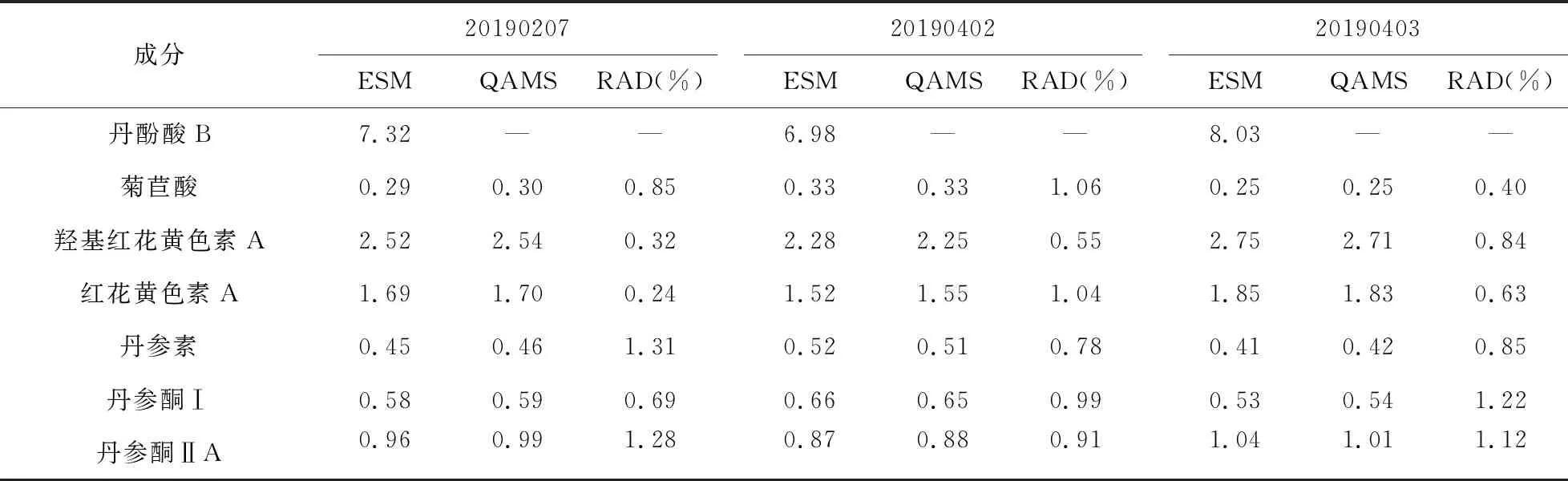
表7 各成分含量测定结果(mg/g)
3 讨论
3.1 供试品溶液制备方法的确定 萃取溶剂的选择,首先采用甲醇[11]、70%甲醇[10-11]、70%乙醇[12]、95%乙醇处理样品,以前列平胶囊中菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA的提取效果为首选指标,同时兼顾杂质峰影响等因素,结果70%甲醇处理样品时,待测成分综合提取率最佳,杂质峰干扰较小;提取方式的选择,对超声提取[6-7,9-10]和回流提取2种提取方式进行对比,结果超声提取、回流提取所得结果差异不大,考虑到样品处理的便捷性,选用超声提取处理样品;提取时间的选择,经试验,确定超声提取30 min效果最佳。
3.2 色谱条件流动相的选择 对甲醇-水[6,11-12]和乙腈-水[7-9]流动相系统进行考察,结果发现,甲醇-水流动相体系检测时,基线漂移严重,同时检测时间过长;乙腈-水流动相体系检测基线平稳,检测用时理想,不足之处是丹酚酸B因含量高,色谱峰出现平头峰和拖尾现象,峰形对称度不符合要求,考虑加入酸类调节。经试验,最终确定采用乙腈-0.4%磷酸水溶液[7,9-10]为流动相进行梯度洗脱,前列平胶囊中待测7种成分的色谱峰峰形对称,与相邻色谱峰均能有效分离。
本文首次采用HPLC-QAMS法对前列平胶囊中菊苣酸、羟基红花黄色素A、红花黄色素A、丹参素、丹酚酸B、丹参酮Ⅰ和丹参酮ⅡA 7种成分的含量进行了同时测定,建立了前列平胶囊多指标成分同时定量的方法,所建立的方法操作便捷,结果准确可靠,对前列平胶囊质量标准提升和全面评价其产品质量,指导药品生产企业优化生产工艺过程控制参数,完善原药材内控标准,确保产品质量的稳定性提供了参考依据。采用高效液相色谱指纹图谱结合多成分质量控制对前列平胶囊进行更加全面的质量评价还在进一步的研究中。
猜你喜欢
农业与技术(2022年11期)2022-11-19
云南化工(2022年7期)2022-07-31
中西医结合心脑血管病杂志(2022年12期)2022-07-07
山东第一医科大学(山东省医学科学院)学报(2022年3期)2022-04-12
现代农村科技(2022年2期)2022-03-04
河北农业(2021年6期)2021-07-17
亚太传统医药(2021年5期)2021-05-24
Medicinal Plant(2020年2期)2020-05-14
医学研究杂志(2015年2期)2015-06-10
爱你(2015年6期)2015-04-20
