
沙门氏菌噬菌体LPST144尾纤维gp38的序列分析及其结合活性
2021-01-20 08:16杨其乐丁一峰聂若男李亚萌王小红
食品科学 2021年2期
杨其乐,丁一峰,张 宇,聂若男,李亚萌,王 佳,王小红
(华中农业大学食品科学技术学院,湖北 武汉 430070)
沙门氏菌是导致食源性疾病的主要病原菌,在全球范围内,每年约造成9 380万 例肠胃炎疾病,以及15.5万 人死亡[1]。沙门氏菌是一类革兰氏阴性短杆菌,需氧或兼性厌氧,肠杆菌科沙门氏菌属,由肠炎沙门氏菌种和邦戈沙门氏菌种组成[2-3]。沙门氏菌分布广泛,菌型繁多,目前使用标准Kauffman-White方案已鉴定超过2 600 种血清型,而大多数血清型能够在包括人类在内的多种动物宿主中寄生,其中肠炎沙门氏菌、鼠伤寒沙门氏菌是导致人畜共患病的两种主要血清型[4]。近年来,抗生素的大量使用导致了耐药性沙门氏菌的出现,耐药沙门氏菌的强毒力、高死亡率引起了广泛关注[5]。因此,为预防沙门氏菌对人类健康以及经济发展造成重大危害,对其进行有效的安全监控和监督、建立快速准确的检测方法具有重要意义。
噬菌体作为一种细菌病毒,能够特异性吸附、侵染细菌并完成自我复制[6],最早发现于一个世纪以前,用于对分子生物学基本原理的探究[7]。在自然环境中,噬菌体几乎无处不在,海洋、土壤、深海喷口以及饮用水、食物中都存在噬菌体。同时,噬菌体也是地球上数量最多的生物体,其数目约为1030~1031,且具有多样性[8]。噬菌体侵染细菌,完成其生命周期的第一步是特异性识别并结合细菌表面受体,其主要识别的细菌表面受体为脂多糖和细菌表面蛋白上特定的残基[9],噬菌体颗粒中承担这一重要功能的蛋白被称为受体结合蛋白(receptor binding proteins,RBPs),存在于噬菌体尾纤维或者尾刺的末端[10-12]。由于RBPs介导的宿主识别是高度特异性的,因此,可以利用RBPs作为分子识别元件进行宿主菌的特异性检测技术研究[13-14]。
目前报道较多的噬菌体RBPs有:T4 gp37、gp12;T2 gp38;P22 gp9和T7 gp17[15-16]。一般而言,RBPs具有如下特点:具有模块化性质,N端连接噬菌体粒子主体,序列同源性较高,C端为受体识别区决定宿主范围,序列相似度较低;许多已知的RBPs受体识别区富含β结构,通常会形成三聚体且结构稳定[16-17];许多尾刺蛋白还具有水解活性,目前已知的具有水解活性的尾刺蛋白已被鉴定有215 个,这些RBPs能够降解脂多糖、磷壁酸等生物聚合物,为噬菌体进入细胞膜注入DNA扫清道路[17];一个噬菌体可以有多个RBPs,例如T4噬菌体具有短尾纤维(gp12)和长尾纤维(gp37)[18],且在吸附过程中承担着不同的功能,T4噬菌体与宿主菌靠近后,长尾纤维(gp37)识别连接细菌表面一级受体,此时的结合可逆,随后T4噬菌体在细菌表面依靠长尾纤维移动,当周围有2~3 个一级受体并与之相连后,信号传递导致底板构象改变,释放短尾纤维(gp12),其不可逆地结合宿主二级受体,最后尾部收缩,注入噬菌体DNA[19]。由于RBPs具有以上特点,将其用作检测探针有以下优点:易于大量获得;RBPs具有模块化的性质,可将受体结合区分离,改造优化,可将具有不同宿主范围的RBPs融合表达,扩大宿主范围[20-21];与易发生重组和突变的完整噬菌体粒子相反,克隆原性在生产过程中保持不变,性质稳定,更适于商业化[16,22]。同时RBPs的一些性质也导致了研究的困难:首先,RBPs序列多样性大,受体识别区序列相似性低,且一种噬菌体可能同时具有一种或者一种以上的RBPs[23],这些特点导致生物信息学方法预测的困难[24];其次,了解噬菌体RBPs的结构能极大促进噬菌体识别机制理论的发展,而尾纤维的柔性结构导致结晶困难,因此对结构研究和识别机制理论的揭示和发展造成了巨大的挑战[25]。
本研究分析实验室前期分离得到的1 株沙门氏菌噬菌体LPST144尾纤维gp38的理化性质、遗传进化关系和二级结构,旨在完成尾纤维基因orf38异源表达纯化以及其特异性结合活性的验证,为后续开发基于噬菌体RBPs为分子探针建立沙门氏菌检测方法奠定实验基础。
1 材料与方法
1.1 材料与试剂
噬菌体LPST144由实验室前期分离保存,为1 株鼠伤寒沙门氏菌(Salmonella typhimuriumATCC13311)烈性噬菌体,短尾科,T7类噬菌体;已完成其基因组测序,GenBank登录号为MN252582;鼠伤寒沙门氏菌ATCC13311(S. typhimuriumATCC13311)、鼠伤寒沙门氏菌ATCC14028(S. typhimuriumATCC14028)、鼠伤寒沙门氏菌ST-8(S. typhimuriumST-8)、鼠伤寒沙门氏菌UK-1(S. typhimuriumUK-1)、肠炎沙门氏菌SJTUF10978(S. enteritidisSJTUF10978)、肠炎沙门氏菌SJTUF10984(S. enteritidisSJTUF10984)、肠炎沙门氏菌10960(S. enteritidis10960)、金黄色葡萄球菌6538(Staphylococcus aureus6538)和大肠杆菌T10(Escherichia coliT10)为实验室保藏菌种;沙门氏菌外膜蛋白Ompc为实验室异源表达纯化得到。
根据噬菌体LPST144基因组注释结果,设计上下游引物扩增orf38基因序列,引物序列如下:orf38F:5’-CGGGATCCCGATGGCACTTAAATT-3’,orf38R:5’-CCGCTCGAGCGGATAGAAGTAGTGAAT-3’(下划线分别代表BamH I和XhoI酶切位点),由生工生物工程(上海)股份有限公司合成。
限制性内切酶、T4 DNA连接酶、聚会酶链式反应产物回收试剂盒 宝日医生物技术(北京)有限公司;质粒pSmart-I、E.coliBL21 美国Addgene公司;HisPur Ni-NTA Resin蛋白纯化树脂 美国Thermo Fisher公司;BCA蛋白定量试剂盒 默克生命科学;鼠抗组氨酸抗体、山羊抗鼠抗体 北京中杉金桥生物技术有限公司;脱脂奶粉 美国BD-Difco公司;9018型酶标板 美国康宁公司。
1.2 仪器与设备
5417R小型台式冷冻离心机 贝克曼库尔特公司;BSO-130011A2超净台 苏净集团苏州安泰空气技术有限公司;HNY-2102C全温振荡培养箱 武汉瑞华仪器设备有限责任公司;M200 PRO酶标仪 瑞士Tecan公司。
1.3 方法
1.3.1 噬菌体LPST144 gp38序列理化性质、二级结构和遗传进化分析
通过在线工具ProtParam(https://web.expasy.org/protparam/)预测gp38序列的理化性质,包括等电点、不稳定系数和亲水系数;使用软件DNAMAN作多序列比对;使用NCBI CDD和Pfam数据库预测序列保守结构域;通过在线工具PredictProtein(https://www.predictprotein.org/)预测gp38序列的二级结构;利用NCBI BLAST工具查找GenBank数据库中与gp38具有相似性的序列,并使用MEGA7绘制进化树(Tree method:Maximum likelihood;Max Seq Difference:0.85;Distance:Grishin)。实验涉及到的尾纤维同源序列包括:LPST144(QEP53513);BP12A(YP_009303689);phiSG-JL2(YP_001949790);SH2(YP_009289339);SH1(YP_009289339);phiIBBPF7A(YP_004306354);Pf-10(YP_009145638);Ebrios(AVJ51925);Kvp1(YP_002308420);Patroon(QBQ72909);fPS-21(SOO46777);fPS-9(SOL37536);fPS-19(SOO46422);fPS-89(SOO46625);vB_KpnP_NahiliMali(QEG13382.1);fPS-10(SOO46575.1);vB_KpnP_Sibilus(QEG12954.1);fPS-59(SOO46827);vB_KpnP_Emp27(QEG11854.1);E-2(YP_009226215);2050H2(ASZ79018);phiYeO3-12(NP_052117);T7(AAM43539);13a(YP_002003979);YpsP-G(AFK13534);T3(NP_523342);phiA1122(NP_848304)。
1.3.2 尾纤维基因orf38的克隆以及重组表达载体的构建
以噬菌体LPST144的基因组为模板,设计引物orf38F(5’-CGGGATCCCGATGGCACTTAAATT-3’)、orf38R(5’-CCGCTCGAGCGGATAGA-AGTAGTGAAT-3’)(下划线分别代表BamH I和XhoI酶切位点)扩增目的基因orf38。使用限制性内切酶BamH I和XhoI酶切扩增产物和质粒pSmart-I,参照TaKaRa公司的BamH I和XhoI说明书,配制酶切体系并选择最佳酶切条件。按照QIAquick Gel Extraction Kit操作说明书对酶切产物进行回收,根据T4 DNA连接酶的说明书,将目的基因与质粒pSmart-I的双酶切产物进行连接,构建重组表达载体,然后转化BL21感受态细胞。使用质粒提取试剂盒提取重组质粒后用EcoR I和XhoI进行双酶切鉴定,将酶切鉴定正确的重组质粒送至公司测序。
1.3.3 重组蛋白的表达和纯化
挑取阳性单克隆接种于含50 μg/mL卡那霉素的LB培养基中,37 ℃过夜培养。取1 mL转接于100 mL含卡那霉素的LB培养基中,37 ℃培养至OD600nm值为0.6~0.8左右。吸取1 mL未经诱导的菌液作为阴性对照,在剩余的菌液中加入异丙基-β-D-硫代半乳糖苷(isopropyl-β-D-thiogalactoside,IPTG)(终浓度1 mmol/L)诱导,将加入IPTG后的菌液分别按下列条件进行培养:16 ℃培养12 h,37 ℃培养12 h,16 ℃培养4 h以及37 ℃培养4 h。收集不同表达条件的样品,12 000 r/min离心15 min收集菌体。用10 mL的磷酸盐缓冲液(phosphate buffered solution,PBS)重悬菌体,超声破碎细胞(功率200 W),超声时间2 s,间隔2 s,总计40 min,之后12 000 r/min离心10 min收集上清液。分别取10 μL样品进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)验证表达情况。
取上述最佳表达条件下的上清液样品,采用HisPur Ni-NTA树脂亲和层析纯化目的蛋白,取1 mL亲和树脂装柱,镍柱用PBS平衡后,加入破碎后的上清样品过柱,分别使用含有50、300 mmol/L咪唑的PBS洗去杂蛋白,并洗脱目的蛋白。纯化后的目的蛋白透析处理,在0.01 mol/L PBS中,4 ℃过夜去除咪唑。将透析后的蛋白样品利用10 kDa超滤管浓缩,4 000 r/min低温离心30 min,弃去滤出液。将最终截留的蛋白样品分装置于-20 ℃保存,取5 μL进行SDS-PAGE检测。采用BCA蛋白定量试剂盒绘制标准曲线,计算样品中的蛋白浓度。
1.3.4 重组蛋白的活性检测
将宿主鼠伤寒沙门氏菌ATCC13311培养至OD600nm值为0.8~1.2,平板计数,离心收集菌体,用1 mL无菌PBS将菌体悬浮于离心管,终质量分数为0.4%的甲醛灭活,4 ℃过夜;将灭活后的菌体用无菌PBS洗涤4 次,并以无菌PBS调整菌浓度至107CFU/mL,取100 μL宿主菌和其他6 株不同血清型的沙门氏菌(鼠伤寒沙门氏菌ATCC14028、鼠伤寒沙门氏菌ST-8、鼠伤寒沙门氏菌UK-1、肠炎沙门氏菌SJTUF10978、肠炎沙门氏菌SJTUF10984、肠炎沙门氏菌10960)包被于96 孔板,同时包被100 μL 5 μg/mL沙门氏菌外膜蛋白、100 μL 107CFU/mL灭活后的金黄色葡萄球菌6538和大肠杆菌T10以及100 μL PBS对照,4 ℃过夜;加入300 μL 3%脱脂牛奶于37 ℃封阻2 h,用PBST(含0.05%吐温20的PBS)洗涤5 次后每孔加入100 μL 0.05 mg/mL的gp38蛋白(加100 μL PBS作为对照),37 ℃孵育1 h,再依次加入50 μL 0.2 μg/mL鼠抗组氨酸抗体(一抗)、50 μL 0.2 μg/mL山羊抗鼠抗体(二抗),每一步之间用PBST洗涤5 次。然后加入底物3,3’,5,5’-四甲基联苯胺在37 ℃显色15 min,最后加入50 μL 2 mol/L硫酸溶液终止反应,并在450 nm波长处测定吸光度。
1.4 数据处理
实验重复3 次,每次设3 个平行。运用GraphPad Prism软件进行绘图和分析,实验数据采用ANOVA进行Duncan差异分析(P<0.05,差异显著)。
2 结果与分析
2.1 尾纤维蛋白gp38基本理化性质分析
利用ProtParam工具对gp38序列理化性质进行分析,gp38由646 个氨基酸组成,等电点为5.94。不稳定系数为22.37,亲水系数为-0.368。一般而言,不稳定系数小于40表明该蛋白较为稳定,亲水系数为负值表明该蛋白具有一定的亲水性。从图1可见,gp38的氨基酸中含量最高的是丙氨酸(A),占全部氨基酸的11.5%,甘氨酸(G)、苏氨酸(T)、缬氨酸(V)、精氨酸(R)和天冬酰胺(N)含量较高,占比在6.5%~9.4%之间,含量最低的是半胱氨酸(C),占全部氨基酸的0.5%。非极性氨基酸占比为39.8%,极性氨基酸的占比较高为60.2%,其中带正电荷的极性氨基酸(K+R+H)有82 个,占12.7%,带负电的极性氨基酸(D+E)有74 个,占11.4%,其余的极性氨基酸为233 个,占36.1%。
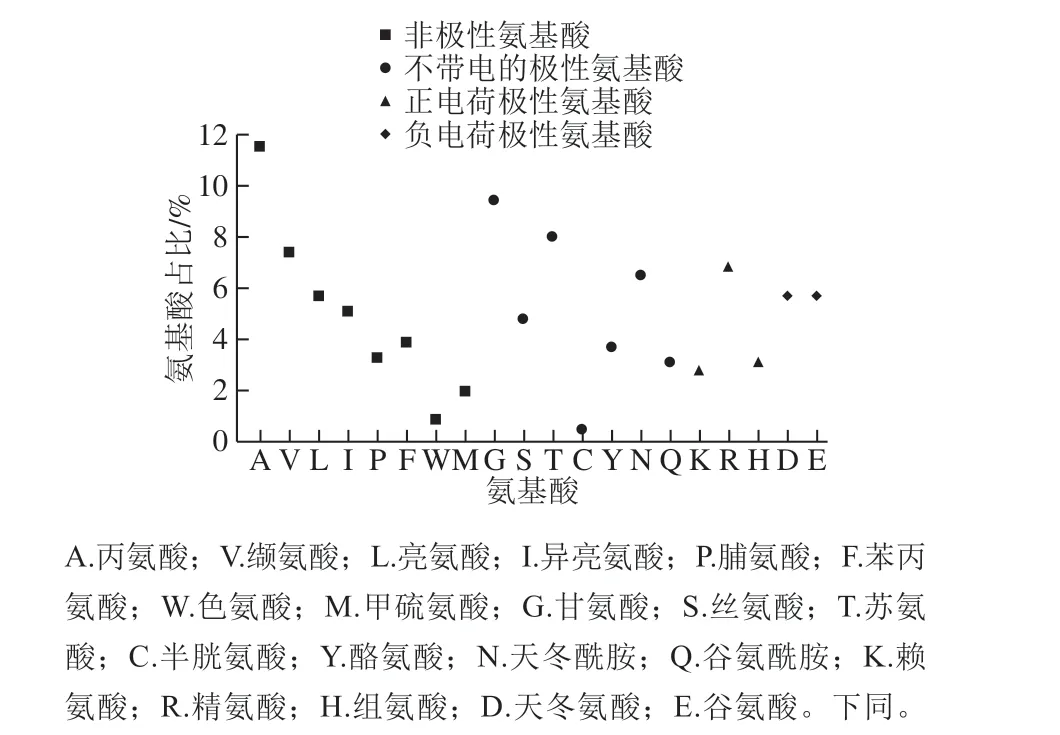
图1 噬菌体LPST144尾纤维gp38氨基酸组成Fig. 1 Amino acid compositions of phage LPST144 tail fiber gp38
2.2 尾丝蛋白gp38的遗传进化分析
将噬菌体LPST144 gp38的氨基酸序列与NCBI nr数据库做BLAST比对,根据比对结果,选择相似度较高的序列用于后续的进化分析。如图2所示,噬菌体LPST144与噬菌体BP12A单独聚为一簇,相似度最高、亲缘关系最近,而与其他噬菌体T7、T3等亲缘关系明显较远,表明噬菌体LPST144和BP12A的尾纤维在序列上与nr数据库中其他噬菌体的尾纤维具有明显的差异。
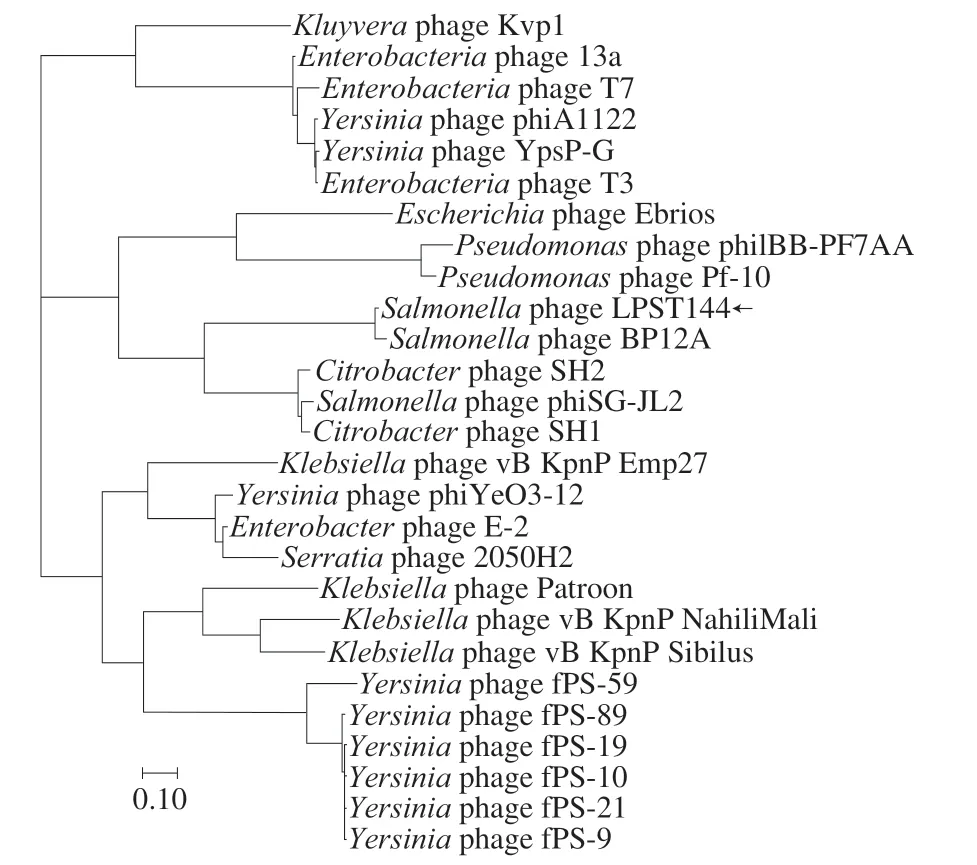
图2 噬菌体LPST144尾纤维gp38的遗传进化分析Fig. 2 Genetic evolution analysis of phage LPST144 tail fiber gp38
根据图2结果,选择亲缘关系不同的8 种噬菌体尾纤维(表1)进一步比对分析。如图3A所示,序列比对结果显示N端保守性较好,同时在该区域预测到两个保守结构域,分别是:1~261位尾纤维蛋白,CDD数据库索引号为cl28018;279~331位噬菌体尾部领状区域,Pfam数据库索引号为pfam07484;而C端序列变异度较大。据文献报道,噬菌体尾纤维的N端通常与噬菌体主体连接,C端负责结合宿主菌,宿主菌表面受体的多样性导致C端序列的多样性[26]。前期工作中鉴定了T7与LPST144同属于Teseptimavirus属,T7噬菌体的尾纤维gp17与噬菌体LPST144的尾纤维gp38的氨基酸序列相似度为41%,覆盖率为54.21%。Garcia-Doval等[27]于2012年对T7噬菌体尾纤维gp17 C端377~533位进行了结构解析,结果表明T7噬菌体gp17最后84 个氨基酸(470~553位)由8 个β-折叠结构(B~I)和无规卷曲组成,形成4 个主要环状结构(BC、DE、FG和HI环),构成T7噬菌体尾纤维尖端结构域的一个单体,是识别和结合宿主菌受体的关键序列。因此将这一部分区域进一步比对分析,结果如图3B所示,总体来看,LPST144与BP12A尾纤维序列匹配非常好,在这一区间内仅有两个碱基的差异,而与phiSGJL2、SH1、SH2和T7、YpsP-G、T3、phiA1122,分为3 组,组内各自匹配度较好,验证了图2进化分析的结果,另一方面,由于各组之间序列差异大,未找到整体的核心保守区。
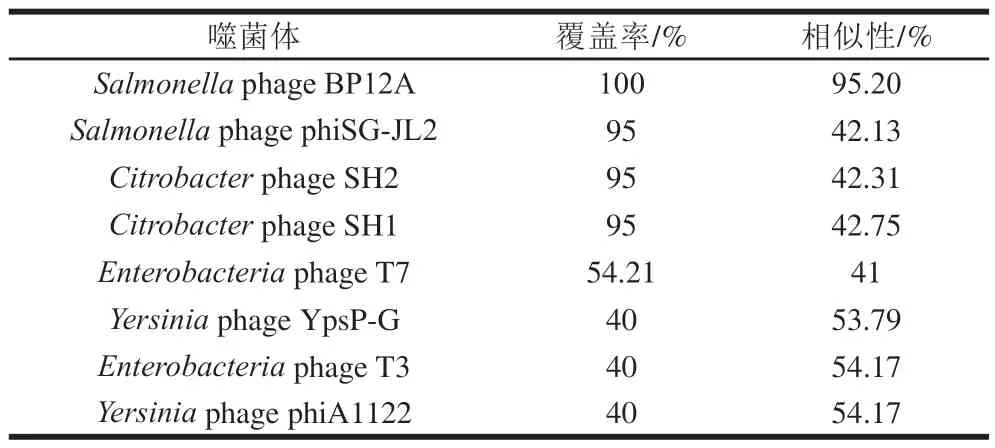
表1 噬菌体LPST144尾纤维gp38的同源性分析Table 1 Homology analysis of phage LPST144 tail fiber gp38
噬菌体T7 gp17的C末端富含β-折叠的区域是受体识别结合关键区,目前已知其520位的天冬氨酸(D)突变为谷氨酸(E)后可裂解E. coliB,544位的缬氨酸(V)突变为丙氨酸(A)后可裂解E. coliK12[28],高同源性噬菌体phiA1122尾纤维523位亮氨酸(L)突变为丝氨酸(S)后,其宿主谱也发生了改变[29](图3B下划线标记处)。通过分析比较LPST144 gp38 C末端的二级结构,发现同样存在大量的β-折叠结构,推测这些富含β-折叠的区域可能与宿主表面受体的识别和结合有关。不同的是T7 gp17的金字塔结构和顶端结构之间由1 个α-螺旋结构连接,顶端结构由8 个β-折叠和无规卷曲结构组成,gp38的C末端由12 个β-折叠(a~n)结构组成,紧接的是1 个α-螺旋结构(o)。T7 gp17具有2 个半胱氨酸(C),不形成二硫键,gp38具有3 个半胱氨酸(C),预测结果显示同样未形成二硫键。另一方面,前期工作中已经鉴定了LPST144与BP12A属于两个不同的物种(未发表),理论上两者的宿主范围会有一定的差异,其gp38 C末端与噬菌体BP12A尾纤维C末端高度相似,仅有2 个氨基酸(533位和612位)的差异(图3B方框标记位点),推测这2 个位点可能是受体识别和结合的关键位点。

图3 噬菌体LPST144尾纤维gp38序列比对分析Fig. 3 Amino acid sequence alignment analysis of phage LPST144 tail fiber gp38
2.3 尾纤维基因orf38的克隆以及重组表达载体pSmart-I-gp38的构建
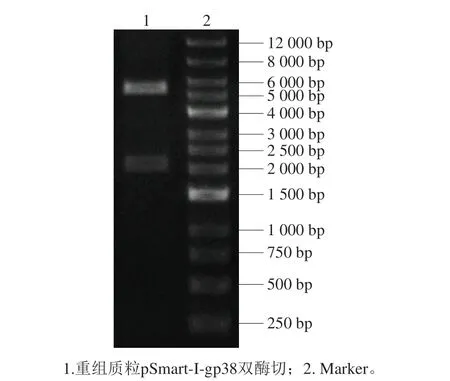
图4 重组质粒pSmart-I-gp38双酶切电泳图Fig. 4 Electrophoresis profile of recombinant plasmid pSmart-I-gp38 after double enzyme digestion
以噬菌体LPST144的基因组序列为模板,设计上下游引物扩增目的基因,使用限制酶BamH I和Xho I酶切扩增产物和pSmart-I空质粒,使用T4连接酶进行连接,构建重组表达载体pSmart-I-gp38。采用Xho I和EcoR I对重组表达载体进行双酶切鉴定,结果如图4所示,酶切产物中包括目的条带(约2 100 bp)和载体条带(约5 400 bp)。同时根据重组质粒的测序结果,表明基因orf38正确插入载体pSmart-I中,重组表达载体pSmart-I-gp38构建成功。
2.4 重组蛋白的表达和纯化
重组表达载体pSmart-I-gp38转化感受态大肠杆菌BL21后,使用IPTG诱导表达,并探究在不同诱导条件下的表达效果。从图5A可知,在80 kDa左右出现预期目的条带,目的蛋白为82.89 kDa。另外,37 ℃诱导4 h上清蛋白表达效果最佳,目的条带明显,杂带浅,因此确定选择37 ℃诱导4 h,并选择上清蛋白作为目的蛋白源进行纯化。重组蛋白纯化结果如图5B所示,可知在75~100 kDa之间出现目标条带,经BCA试剂盒定量测定其质量浓度为0.5 mg/mL。
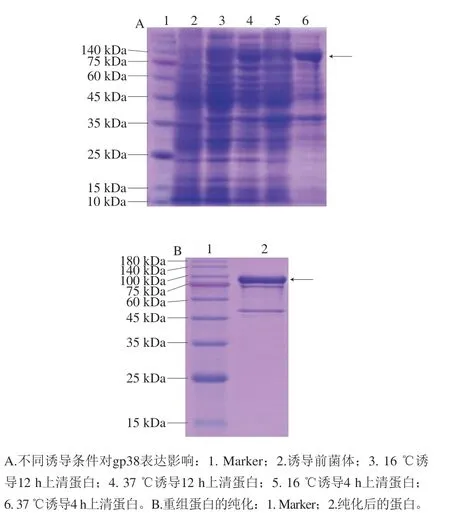
图5 gp38蛋白的表达与纯化Fig. 5 SDS-PAGE profiles of expressed and purified gp38 protein
2.5 重组蛋白gp38的活性
将宿主沙门氏菌ATCC13311加入酶标板过夜,使其附着在酶标板上;脱脂牛奶封阻后加入gp38蛋白进行孵育;然后依次加入一抗、二抗和底物进行显色,加入浓硫酸终止反应,检测重组蛋白的结合活性。结果表明(图6),实验组(加gp38蛋白)的吸光度为0.94±0.02,极显著高于阴性对照(PBS代替gp38蛋白)的吸光度0.17±0.01,表明gp38蛋白具有受体结合活性。为进一步验证gp38蛋白的结合特异性,分别用大肠杆菌T10、沙门氏菌外膜蛋白、金黄色葡萄球菌6538和PBS代替宿主沙门氏菌ATCC13311作为对照组,并测试其他6 株不同血清型的沙门氏菌。如图7所示,对照组吸光度分别为0.58±0.03、0.56±0.01、0.59±0.03、0.53±0.005,实验组(包括宿主在内的7 株沙门氏菌组)的吸光度较各对照组明显更高,且gp38蛋白与大肠杆菌T10、金黄色葡萄球菌6538和外膜蛋白的结合较PBS无明显差异,表明LPST144尾纤维gp38具有特异的受体结合活性。
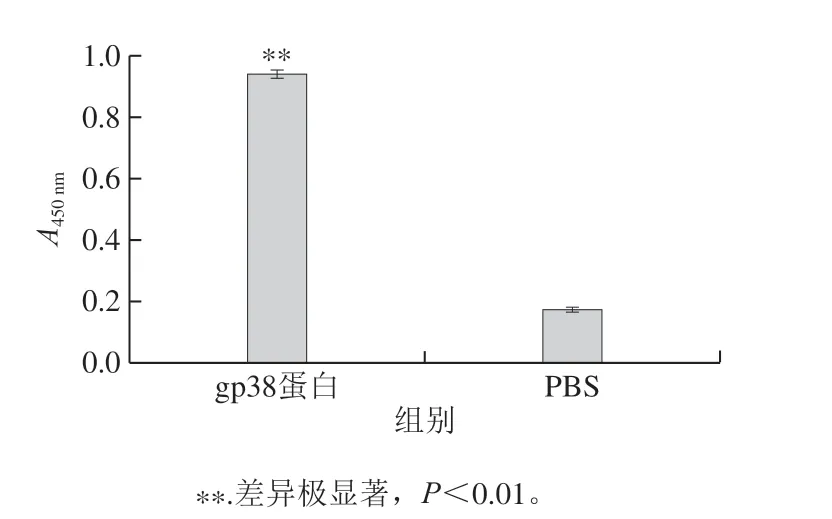
图6 重组蛋白gp38对宿主菌的结合活性Fig. 6 Binding activity of recombinant protein gp38 to host cells
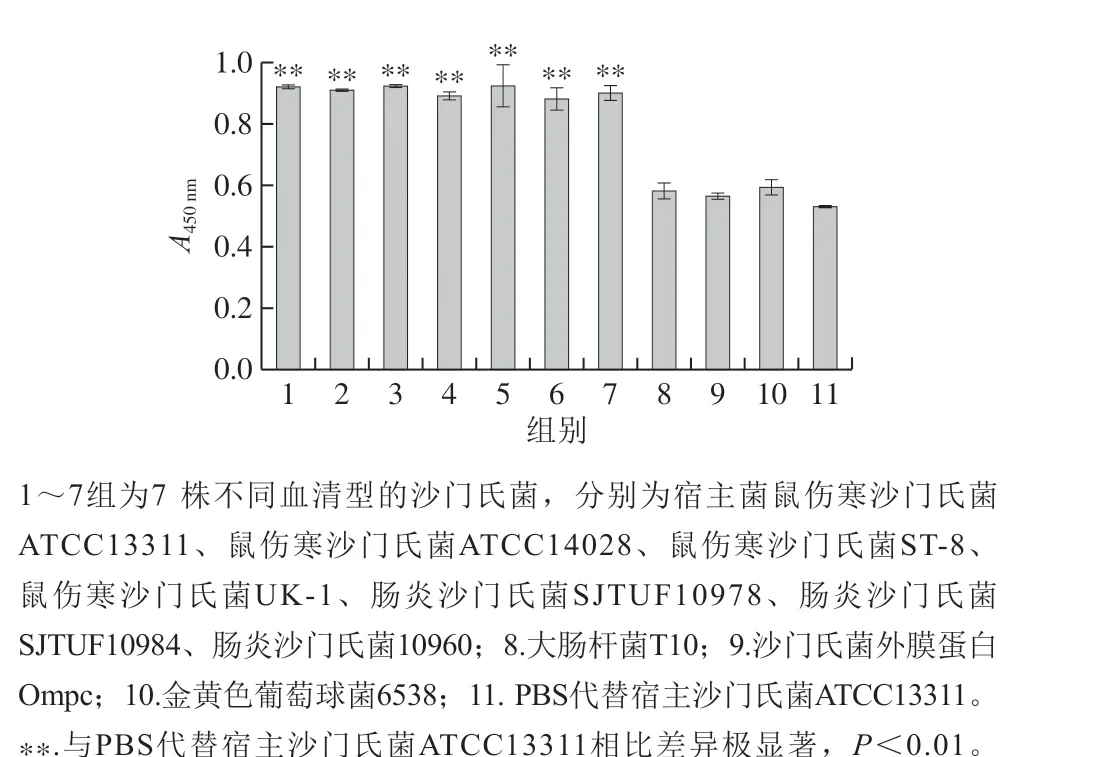
图7 重组蛋白gp38的特异性结合活性Fig. 7 Specific binding activity of recombinant protein gp38
3 讨 论
沙门氏菌噬菌体LPST144是本实验室前期分离得到的1 株短尾科噬菌体,其吸附时间短,9 min便达到最大吸附量77.57%,具有用于沙门氏菌快速检测方法开发的潜力。通过测序分析,鉴定其与T7噬菌体同属于Teseptimavirus属,预测了其尾纤维gp38(发表中),本研究对其尾纤维进行了进一步的序列分析和活性鉴定。目前已知晶体结构信息的尾纤维或尾刺数量非常有限,现已有报道的RBPs晶体结构有短尾科噬菌体P22的尾刺gp9[30]、T4噬菌体的gp37[31]以及T7噬菌体的尾纤维gp17[27],通过对LPST144 gp38进行序列分析,发现LPST144尾纤维gp38与T7噬菌体尾纤维gp17的N端序列具有较好的相似性,C端相似性却极差。并通过遗传进化、多序列比对和二级结构分析,发现其满足RBPs的以下特征:具有模块化的性质,N端保守性强而C端多样性大;C端富含β折叠结构;序列相似性低。将其C端序列与GenBank数据库比对,发现仅与BP12A的尾纤维具有较好的相似性,而与其他序列同源性不高。BP12A为1 株沙门氏菌噬菌体,GenBank登录号为KM366096,经基因组比较鉴定,其与噬菌体LPST144是2 个不同的物种,理论上宿主范围存在一定差异,而两者的尾纤维C端仅有两个位点(533位和612位)的差异,因此推测这两个位点可能是受体结合关键氨基酸。由于PDB数据库中缺乏能够满足同源建模要求的结构模板,而采用从头计算的方法预测得到的三维结构未能通过评估,因此未能得到gp38三维结构,而LPST144的尾纤维与其他噬菌体(除噬菌体BP12A)的尾纤维几乎没有相似性,因此未来有必要对其空间构象进行解析,为扩宽其宿主谱提供指导依据。
目前报道的常用于沙门氏菌检测的RBPs有短尾科噬菌体P22的尾刺gp9[17]和肌尾科噬菌体S16的尾纤维gp38[32],本研究对短尾科噬菌体LPST144的尾纤维进行异源表达,验证了尾纤维gp38的特异性结合能力。在异源表达实验中,最初使用的表达质粒为pET28b,由于表达产物溶解性差形成包涵体,于是在后续的实验中采用了携带His标签和SUMO标签的促融表达质粒pSmart-I。实验结果表明,增加SUMO标签能显著提高目的蛋白的溶解性。通过考察确定最佳的诱导条件为37 ℃诱导4 h,纯化后测定其蛋白质量浓度为0.5 mg/mL。在目的蛋白结合能力的实验中,gp38蛋白加入后的吸光度较阴性对照明显提高,表明了gp38蛋白对于宿主菌具有较强的结合能力。在验证其特异性结合能力的实验中,发现gp38对于宿主菌以及其他6 株受试的不同血清型沙门氏菌较大肠杆菌T10、沙门氏菌外膜蛋白、金黄色葡萄球菌6538和PBS代替宿主菌对照组的吸附明显更强,而包被大肠杆菌T10、沙门氏菌外膜蛋白和金黄色葡萄球菌6538组与PBS代替宿主菌对照组无显著性差异,表明了gp38蛋白的结合特异性,相比于常见的酶联免疫吸附实验,实验结果中对照组的吸光度偏高,推测这种现象的产生可能是由于gp38重组蛋白N端的SUMO标签未切割所造成,产生了一定的非特异性结合。通常情况下,重组表达蛋白的活性受到表达条件(温度、pH值等)、表达菌株和表达过程中重组蛋白是否正确折叠等诸多因素的影响,因此,后期还有待对这样因素加以考虑并优化条件,针对这一问题进行改进。通常RBPs的宿主范围会与完整噬菌体颗粒相当或者更为广泛,沙门氏菌血清型众多,目前已分离鉴定2 600多种,同时可大规模测定gp38结合的宿主范围,以明确检测的对象范围。已知目前报道的用于沙门氏菌的分子探针P22 gp9和S16 gp38对部分其他血清型鼠伤寒沙门氏菌作了测试,而鼠伤寒沙门氏菌ATCC13311均未被测试,一般来讲,由于RBPs的序列差异大,且沙门氏菌血清型众多(目前已发现超过2 600 个沙门氏菌血清型),能够结合的宿主范围差异也会很大。另一方面,本实验是一个初步的功能验证实验,其目的在于证明gp38具有RBPs特异性结合活性,具有作为检测探针的潜力,后期课题组将进一步优化gp38重组表达和活性研究并开发检测方法。一般来说检测灵敏度是针对检测体系的建立,因此目前未能进行灵敏度的比较。
4 结 论
迄今为止,沙门氏菌感染仍然是世界范围内主要的公共卫生问题,每年沙门氏菌的感染事件给人类健康以及经济社会造成重大负担,噬菌体作为细菌的天敌,能够特异性吸附并侵染宿主菌,给沙门氏菌的检测提供了新的思路。本研究对短尾科沙门氏菌噬菌体LPST144的尾纤维基因orf38进行研究,预测了其序列和结构信息,通过异源表达纯化,验证了gp38具有特异性结合宿主沙门氏菌以及实验中其他受试沙门氏菌的能力,为开发基于噬菌体RBPs为分子探针建立沙门氏菌的检测方法奠定了实验基础,同时也为理解噬菌体与宿主菌的相互作用提供了依据。
猜你喜欢
动物医学进展(2022年9期)2022-09-06
中国食品学报(2022年6期)2022-07-19
昆明医科大学学报(2022年2期)2022-03-29
植物保护(2021年4期)2021-11-12
中国饲料(2021年17期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
科学(2020年3期)2020-11-26
当代水产(2020年3期)2020-06-15
科学24小时(2020年4期)2020-05-14
小星星·阅读100分(高年级)(2015年11期)2015-11-28
