
Oxidative stress in alcohol-related liver disease
2021-01-14 00:34:24HueyTanEuanYatesKristenLillyAshwinDhanda
World Journal of Hepatology 2020年7期
Huey K Tan, Euan Yates, Kristen Lilly, Ashwin D Dhanda
Huey K Tan, Euan Yates, Kristen Lilly, Ashwin D Dhanda, Hepatology Research Group, Institute of Translational and Stratified Medicine, Faculty of Health, University of Plymouth, Plymouth PL6 8BU, United Kingdom
Huey K Tan, Ashwin D Dhanda, South West Liver Unit, University Hospitals Plymouth NHS Trust, Plymouth PL6 8DH, United Kingdom
Kristen Lilly, Department of Clinical Immunology, University Hospitals Plymouth NHS Trust, Plymouth PL6 8DH, United Kingdom
Abstract Alcohol consumption is one of the leading causes of the global burden of disease and results in high healthcare and economic costs.Heavy alcohol misuse leads to alcohol-related liver disease, which is responsible for a significant proportion of alcohol-attributable deaths globally.Other than reducing alcohol consumption, there are currently no effective treatments for alcohol-related liver disease.Oxidative stress refers to an imbalance in the production and elimination of reactive oxygen species and antioxidants.It plays important roles in several aspects of alcohol-related liver disease pathogenesis.Here, we review how chronic alcohol use results in oxidative stress through increased metabolism via the cytochrome P450 2E1 system producing reactive oxygen species, acetaldehyde and protein and DNA adducts.These trigger inflammatory signaling pathways within the liver leading to expression of pro-inflammatory mediators causing hepatocyte apoptosis and necrosis.Reactive oxygen species exposure also results in mitochondrial stress within hepatocytes causing structural and functional dysregulation of mitochondria and upregulating apoptotic signaling.There is also evidence that oxidative stress as well as the direct effect of alcohol influences epigenetic regulation.Increased global histone methylation and acetylation and specific histone acetylation inhibits antioxidant responses and promotes expression of key pro-inflammatory genes.This review highlights aspects of the role of oxidative stress in disease pathogenesis that warrant further study including mitochondrial stress and epigenetic regulation.Improved understanding of these processes may identify novel targets for therapy.
Key words: Alcohol-related liver disease; Alcoholic hepatitis; Oxidative stress; Reactive oxygen species; Antioxidants; Epigenetics; Mitochondrial stress
INTRODUCTION
Europe has the highest per capita alcohol consumption and alcohol-related loss of disability adjusted life years globally[1].The European Union is the heaviest drinking region of the world, with 11 liters of pure alcohol drunk per adult each year[1].In the last decade, this trend continues to rise in central and northern Europe.Alcohol is a dose-related risk factor for more than 200 diseases[1].It causes 5.9% of all deaths globally and more than 25% of deaths in the age group 20-39 years[1].
Heavy alcohol use is the cause of alcohol-related liver disease (ALD).Therefore, it is not surprising that the incidence of ALD is on a rising trajectory[2].The scale of ALD is estimated to burden the United Kingdom national health system with health-related costs of over £3.5 billion per year[2].In Europe, the cost of treating ALD is estimated to be €17 billion, together with €5bn spent on treatment and prevention of harmful alcohol use and alcohol dependence[3].Worldwide, alcohol-related liver cirrhosis deaths account for approximately 10% of all alcohol-attributable deaths resulting in the loss of 22.2 million disability-adjusted life years annually[4].
The ALD spectrum ranges from simple steatosis to steatohepatitis, fibrosis, and cirrhosis.While alcohol-related cirrhosis is no longer considered a completely irreversible condition, no effective anti-fibrotic therapies are currently available.Cirrhosis can be divided into compensated and decompensated stages, with differentiating clinical features and prognosis.The median survival of patients with compensated liver disease is approximately 6.5 years but only 2.5 years in those with decompensated cirrhosis[4].Once a complication of cirrhosis develops, the 5-year survival rate decreases to less than 20%[4].
Alcoholic hepatitis (AH) is an acute inflammatory condition that occurs on the background of ALD.Severe AH has a mortality rate of 30% within 3 mo[5]but even non-severe AH has a significant 7% mortality within 3 mo[6].The established treatment for AH is corticosteroids, which improve short-term survival but do not affect longterm survival[5].
The molecular mechanisms underlying ALD pathogenesis are complex and have not been fully elucidated.However, there is emerging evidence that oxidative stress plays a role in mediating the inflammatory response and in directly causing liver damage.Oxidative stress represents the body’s imbalance in the production and the elimination of reactive species (including reactive oxygen and nitrogen species) as well as decreased production of antioxidants[7].Here, we review the role of oxidative stress in ALD focusing on its effect on mitochondrial stress, cell signaling and epigenetic regulation.
LITERATURE SEARCH
Comprehensive searches of MEDLINE, EMBASE, PubMed and TRIPS from their commencement to June 2019 were conducted.The search strategy included subject headings and keywords related to “alcohol” and “oxidative stress” and “liver”.The reference list of all included studies was screened for eligibility.This review included all study types in humans and animals.Studies published in all languages were considered.One author independently screened titles and abstracts and subsequently reviewed full-texts of retrieved studies for eligibility.
ALCOHOL METABOLISM
Alcohol (ethanol) is metabolized by three major pathways (Figure 1)[7].The primary pathway is initiated by alcohol dehydrogenase (ADH), a NAD+requiring enzyme expressed at high levels in hepatocytes, which oxidizes ethanol to acetaldehyde[7].In a normal liver, acetaldehyde enters the mitochondria and is quickly metabolized to acetate by aldehyde dehydrogenase (ALDH).Acetate is then broken down to carbon dioxide and water for elimination[8].In chronic alcohol users, the ADH/ALDH pathway becomes saturated and reactive aldehydes are produced from the metabolism process such as malondialdehyde-acetaldehyde (MAA), 4-hydroxy-2-nonenal (HNE) and lipid hydroperoxides which can bind to proteins to produce protein adducts[8].
These protein adducts are capable of provoking an immune response.In vitroexperiments showed that the viability of antigen-presenting cells, lymphocytes, and hepatocytes was decreased on incubation with an MAA hen egg lysosome adduct[9].Circulating antibodies against MAA protein adducts were increased in patients with ALD and AH and correlated with the severity of liver injury[9].
The second major pathway to metabolise ethanol is the microsomal ethanol oxidizing system (MEOS), which involves an NADPH-requiring enzyme, the cytochrome P450 enzyme CYP2E1[10], which is induced by chronic alcohol exposure[11,12].The increase of CYP2E1 after alcohol intake is due to stabilization of CYP2E1 rather than to a de novo synthesis[11].The MEOS pathway metabolises ethanol to acetaldehyde by converting NADPH+and O2to NADP and H2O resulting in the generation of reactive oxygen species (ROS).CYP2E1 plays a role in lipid peroxidation, protein oxidation, and protein nitration (Figure 1)[11].It is also known to promote hepatic carcinogenesis by oxidizing DNA in alcohol-exposed rodents[13].
Ethanol metabolism through CYP2E1 not only produces acetaldehyde but also generates ROS including H2O2, hydroxyl (OH-) and carbon centered OH-(Figure 1)[14].These ROS may be neutralized by a potent antioxidant defense system[14].However, chronic alcohol consumption disrupts this system; depletion of mitochondrial glutathione (GSH) is observed in patients with alcohol dependence[15], which impairs hepatocyte tolerance to tumour necrosis factor alpha (TNF-α) resulting in an increased likelihood of cell death[16].ROS increases and activates c-Jun N-terminal kinase (JNK) with consecutive expression of the activator protein 1 (AP-1) transcription factor leading to cellular hyper-regeneration, and lipid peroxidation.Lipid peroxidation products such as malondialdehyde and HNE are generated.HNE can bind to adenosine and cytosine forming highly carcinogenic exocyclic etheno DNA adducts[17].These DNA adducts have been identified in the livers of patients with ALD and other types of liver disease associated with inflammation and oxidative stress like viral hepatitis[18].
The other two most prevalent DNA adducts are N2-ethyldeoxyguanosine (N2-EtdG), and 1,N(2)-propano-2′-deoxyguanosine (PdG).N2-Et-dG is detectable in livers of alcohol-exposed mice and leukocytes of human alcohol misusers[19].PdG, on the other hand, is distinguished by its genotoxic and mutagenic effects which impair DNA replication, thereby triggering cell death.These two major acetaldehyde-DNA adducts also promote carcinogenesis by initiating replication errors and mutations in oncogenes/onco-suppressor genes[19].
A third minor pathway for ethanol metabolism involves catalase, a peroxisomal enzyme (Figure 1)[20], which requires the presence of H2O2, a breakdown product of fatty acids.Catalase located in the peroxisomes of the hepatocyte plays only a minimal role in alcohol metabolism due to low hepatic production of H2O2.Under normal conditions, ADH metabolizes about 75%-80% of the ethanol entering the liver and MEOS the remainder.Hepatic ADH and hepatic catalase activities remain unchanged following chronic alcohol consumption, whereas hepatic MEOS activity strikingly increases and is responsible for the enhanced alcohol metabolism found after chronic alcohol consumption[11,12].

Figure 1 The three major pathways of alcohol metabolism.
MITOCHONDRIAL STRESS
Chronic alcohol consumption results in structural and functional abnormalities in hepatic mitochondria, including enlarged morphology[21,22], mitochondrial DNA (mtDNA) damage[23], reductions in hepatic ATP levels[24]and mitochondrial protein synthesis[25](Figure 2).This can result in hepatocellular apoptosis and associated necrosis[26].Chronic alcohol metabolism and associated mitochondrial dysfunction has been implicated in increasing ROS production and accumulation in hepatic mitochondria.
In humans,in vivomeasurement of ROS is complicated due to their rapid reactions with surrounding molecules[27].Surrogate measures of mitochondrial-derived ROS include urinary isoprostane levels[28,29], NADH delivery to the respiratory chain[30], lipid peroxidation[31,32]and HNE levels and associated adducts[17,33].The greatest indicator of ROS overproduction is the increase in hepatic CYP2E1 levels[32,34-37].The respiratory chain has also been implicated in mitochondrial ROS overproduction in response to chronic alcohol consumption.Excessive levels of reducing equivalents (e.g., NADH), produced by alcohol and ADH entering the mitochondrial respiratory chain, lead to electron transport chain reduction, facilitating superoxide anion formation[38,25].
Cell death can be triggered through ROS-induced release of apoptosis signalregulating kinase 1 (ASK1) (a member of the mitogen-activated protein kinase [MAPK] family), resulting in the cleavage of pro-caspase-3 to active caspase-3, which promotes cellular apoptosis[39-41].Additionally, cytosolic ASK1 activates MAPK kinase 4 and JNK resulting in increased mitochondrial permeability, mediated by SAB protein, and thus hepatocyte cell death[40,41](Figure 2).
Reactive nitrogen species (RNS) also contribute to mitochondrial damage[22,38].Alcohol-mediated overproduction of the superoxide anion can result in the generation of RNS, such as peroxynitrite,viainteraction with nitric oxide, culminating in mitochondrial protein damage[25].Numerous mitochondrial-localized enzymes involved in respiration and cellular energetic processes are inactivated in this way, including NADH dehydrogenase, succinate dehydrogenase, cytochromecreductase and ATP synthase[42].
To limit oxidative damage following alcohol consumption, hepatic mitochondria have various adaptive mechanisms to prevent functional and structural impairments.Uncoupling proteins (UCPs), specifically UCPs 1-3, reduce ROS production by the uncoupling of mitochondrial oxidative phosphorylation[43], a process observed in patients with non-alcoholic fatty liver disease (NAFLD)[44].Furthermore, there is mitochondrial upregulation of enzymatic antioxidants catalase, glutathione transferase and heme oxygenase-1 and a marked increase in GSH[45,46].However, mitochondrial GSH depletion was observed in patients with alcohol dependence and ALD[15,16]suggesting that chronic alcohol exposure downregulates GSH expression.
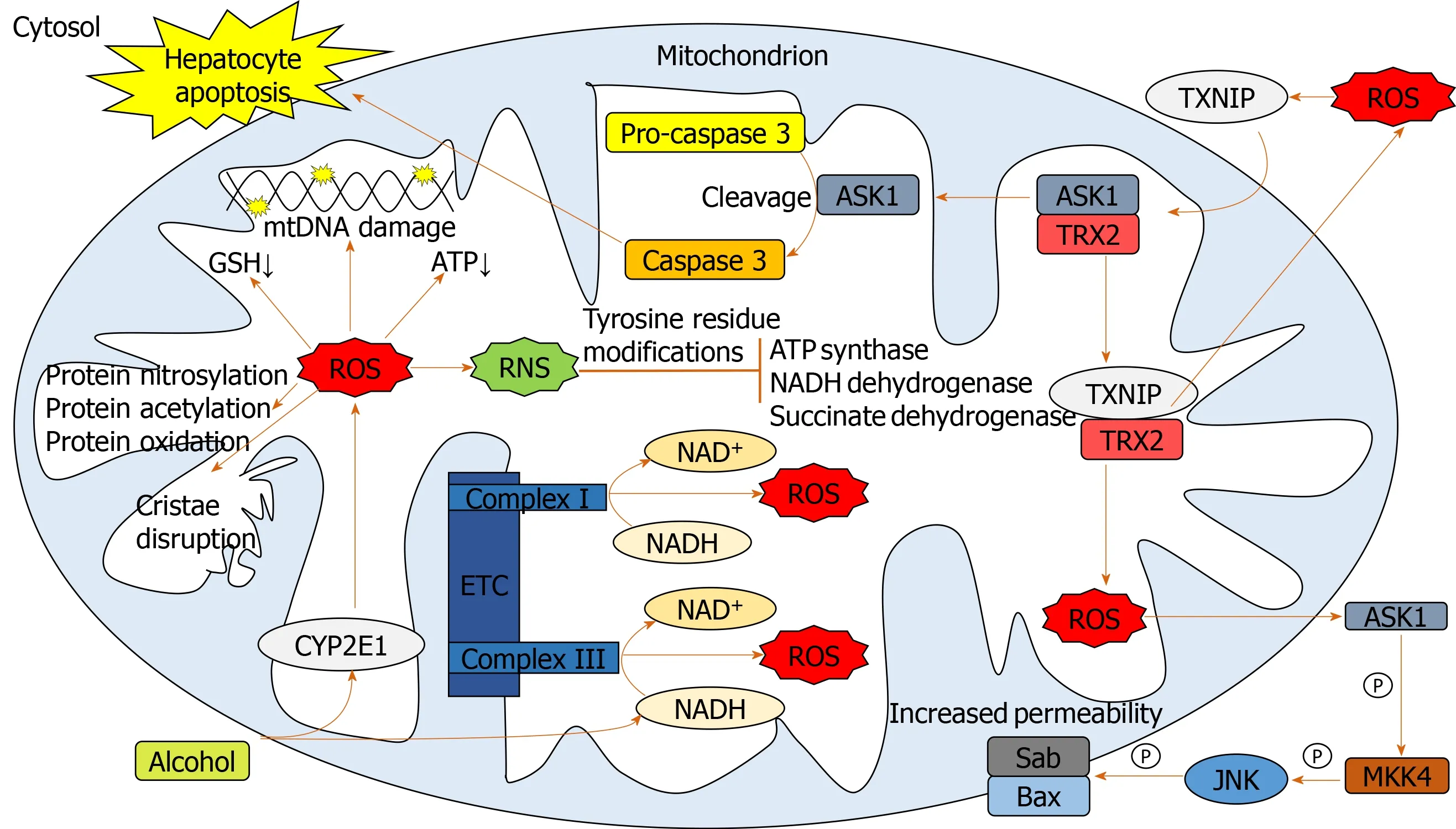
Figure 2 Pathways involved in mediating mitochondrial oxidative stress.
Manganese-dependent superoxide dismutase (MnSOD) detoxifies mitochondrial superoxide[47], but its response to alcohol is poorly documented.Increased mitochondrial localization of MnSOD was associated with more severe forms of ALD[48], which may be mediated by increased hydroxyl radical generation[22].Thus, overexpression of MnSOD may be hepatotoxic rather than hepatoprotective.
S-adenosylmethionine (SAMe) has been implicated in regulating mitochondrial function, following alcohol consumption in a variety of animal models[49].SAMe binds and inactivates the catalytic activity of CYP2E1[50], limiting alcohol-dependent increases in mitochondrial production of superoxide[49].SAMe also increases synthesis and availability of glutathione[51]and maintains mitochondrial respiration rate and mtDNA integrity[38].Although greater SAMe levels have been observed in the serum of ALD patients compared to healthy subjects[52], a reduction in hepatic SAMe levels was observed in patients with AH[53], suggesting the acute inflammatory state leads to hepatic SAMe depletion.SAMe has been evaluated as a treatment for AH in a recent phase 2 randomized controlled clinical trial.SAMe with prednisolone improved 6-mo survival compared to prednisolone treatment alone[54].Although these preliminary results are encouraging, a definitive study has yet to be undertaken.
CELL SIGNALING PATHWAYS
Lipopolysaccharide (LPS) plays a key role in the pathogenesis of ALD, with higher circulating LPS levels in alcohol dependent patients[55,56].In AH, LPS predicts organ failure, mortality[57]and infection[58].Alcohol exposure increases gut permeability, mediating translocation of LPS from the lumen of the intestine to the portal vein into the liver[55].LPS binds to Toll-like receptor 4 (TLR4) expressed on a wide variety of immune and parenchymal cells including Kupffer cells, hepatocytes, endothelial cells and hepatic stellate cells, initiating one of the primary signaling cascades associated with liver damage[59,60].LPS-mediated cell signaling results in transcription of proinflammatory genes through nuclear factor-κB (NF-κB) and interferon regulatory factor 3 DNA binding[59,61].
Upon LPS stimulation of the TLR4 complex, NADPH oxidase (NOX) 4 interacts with the COOH-terminal region of TLR4 resulting in ROS generation in neutrophils and monocytes[62,63], which directly activates NF-κB[62,64].ROS-mediated activation and potential regulation of NF-κB activity occurs by several mechanisms: IκBαphosphorylation; S-glutathionylation of IKKβ; disruption of IκB ubiquitination and degradation; NF-κB inducing kinase (NIK) activation and phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) stimulation[65](Figure 3).ROS both negatively and positively regulates NF-κB, with oxidative stress in the early phase being a positive regulator, compared to a negative regulator in the late phase[66].Diphenyliodonium (DPI), an inhibitor of NOX, used as a pre-treatment in alcohol-fed rats, results in normalized ROS production, and inhibition of TNF-α production in Kupffer cells[59,67].Treatment of alcohol-fed rats with the antioxidant dilinoleoyl-phosphatidylcholine, also inhibited TNF-α production in Kupffer cells and LPS-induced NF-κB activation[68].
Diphenyliodonium and dilinoleoyl-phosphatidylcholine reduce extracellular signalregulated protein kinase (ERK)1/2 activation[67,68].LPS-induced activation of ERK1/2 results in transcription of early growth response protein 1 (Egr-1), involved in binding to the TNF-α promoter and increasing TNF-α expression[59].Egr-1 deficient mice are protected from chronic alcohol-induced liver injury in association with decreased TNF-α messenger RNA (mRNA) levels[69].
LPS activates other MAPKs including p38 and JNK[59], involved in TNF-α production[70].p38 has been implicated in maintaining the stability of TNF-α mRNA[59,71].In response to acute alcohol exposure, the JNK pathway has been associated with increased hepatic mitochondrial ROS production[72], increased JNK phosphorylation and AP-1 binding in monocytes[73].ROS is likely to activate JNK through interaction with upstream MEKK1[65]and by inactivating JNK inhibitor dual specificity protein phosphatase 1[40,74].ROS have also been associated with activation of cytosolic ASK1[40](Figure 3).Clinical trials of ASK1 inhibitors as a treatment for inflammatory liver disease are ongoing with a suggestion of reduced fibrosis in patients with NAFLD[75]but no efficacy seen in AH[76].
ROS-mediated S-glutathionylation results in decreased expression of downstream antioxidants such as MnSOD, catalase and Sestrin3viathe PI3K/AKT pathway[77].Akt has also been implicated in increasing oxygen consumption, resulting in elevated mitochondrial generation of H2O2,facilitating further oxidative damage[78,79].
The net result of these alcohol-induced cell signaling pathways is the increased production of pro-inflammatory cytokines through upregulation of transcription factors such as AP-1 and NFκB.TNF-α, a key pro-inflammatory cytokine, is highly elevated in patients with ALD and AH[80-82], with observed TNF-α gene expression increasing in ALD patients[83].TNF-α induces apoptosis through interaction with TNFα receptor 1 (TNFR1), initiating a cell-death cascadeviaactivation of caspases[84].In ALD, TNF-α-induces mitochondrial peroxidation[55], which is worsened following depletion of GSH[15,85].
TNF-α exacerbates oxidative damage and inflammationviaa positive feedback loop.Through association with TNFR1, TNF-α stimulates the association of complex I[86], which culminates in MAPK activation (JNK, p38 and ERK).Complex I also directly contributes to ROS accumulation through generation of superoxide, capable of causing further oxidative damage and eventual TNF-α, perpetuating the cycle[40,87,88].
Soluble inflammatory mediators including interleukins have been implicated in ALD[60,89,90]and are associated with outcome in patients with AH[91].Elevated serum IL-6 levels have recently been identified as a predictor of mortality in severe AH patients[64].Hepatic upregulation of IL-6 and IL-1β in ALD, results in the differentiation of naïve CD4+cells into IL-17-producing T-helper 17 cells (Th17) (Figure 4), resulting in elevated hepatic and serum levels of IL-17 observed in ALD patients[64,92].IL-17 has a multitude of pro-inflammatory downstream effects, including inducing neutrophil recruitment to the liver; stimulating IL-8 and CXCL1 production by hepatic stellate cells[93]and CXCL4, 5 and 6 expression[92,93].IL-6 and interferon (IFN)-γ are involved in JAK/STAT activation promoting hepatic regeneration[59,94].Conversely, despite upregulation of IL-6 in ALD patients, downregulation of STAT activation has been observed in human monocytes with chronic alcohol exposure[95].

Figure 3 Signaling pathways involved in exacerbating oxidative damage and liver injury.
Inflammasomes propagate IL-1β and IL-18 signals, important in the regulation of hepatic inflammation[94].ROS mediates IL-1β and IL-18 signalingviainflammasome NLRP3 activation[96,97]and inhibition of antioxidant molecules[41](Figure 4).Increased production of IL-1β is critical in Th17 differentiation[64,92,98], while IL-18 activates natural killer T-cells (NKTs) to produce IFN-γ[99].Anti-IL-18 antibodies reduce activation of NF-κB and AP-1, inflammation, liver damage and mortality in animal models[99,100].IL-1β has also been identified as an activator of MAPKs, including p38, JNK, MEKK1 and IKKβ, involved in mediating upregulation of itself and other pro-inflammatory cytokines[101], creating another positive feedback loop.
TRACE ELEMENTS
Trace elements are a group of naturally occurring minerals that are nutritionally fundamental to basic cellular and immunological functions[102].An essential role of the these molecules, including zinc, copper, selenium and manganese, is to act as cofactors of anti-oxidant enzymes, making their role imperative in the context of oxidative stress[103,104].Manganese, copper and zinc are part of the SOD enzyme group that catalyze the breakdown of highly reactive superoxide radicals to H2O2or O2-.Selenium is a component of the active site of glutathione peroxides (GPx), the main function of which is the neutralization of hydrogen peroxide[105].These enzyme systems are crucial in counterbalancing the oxidative stress state and are impaired in chronic liver disease[106].
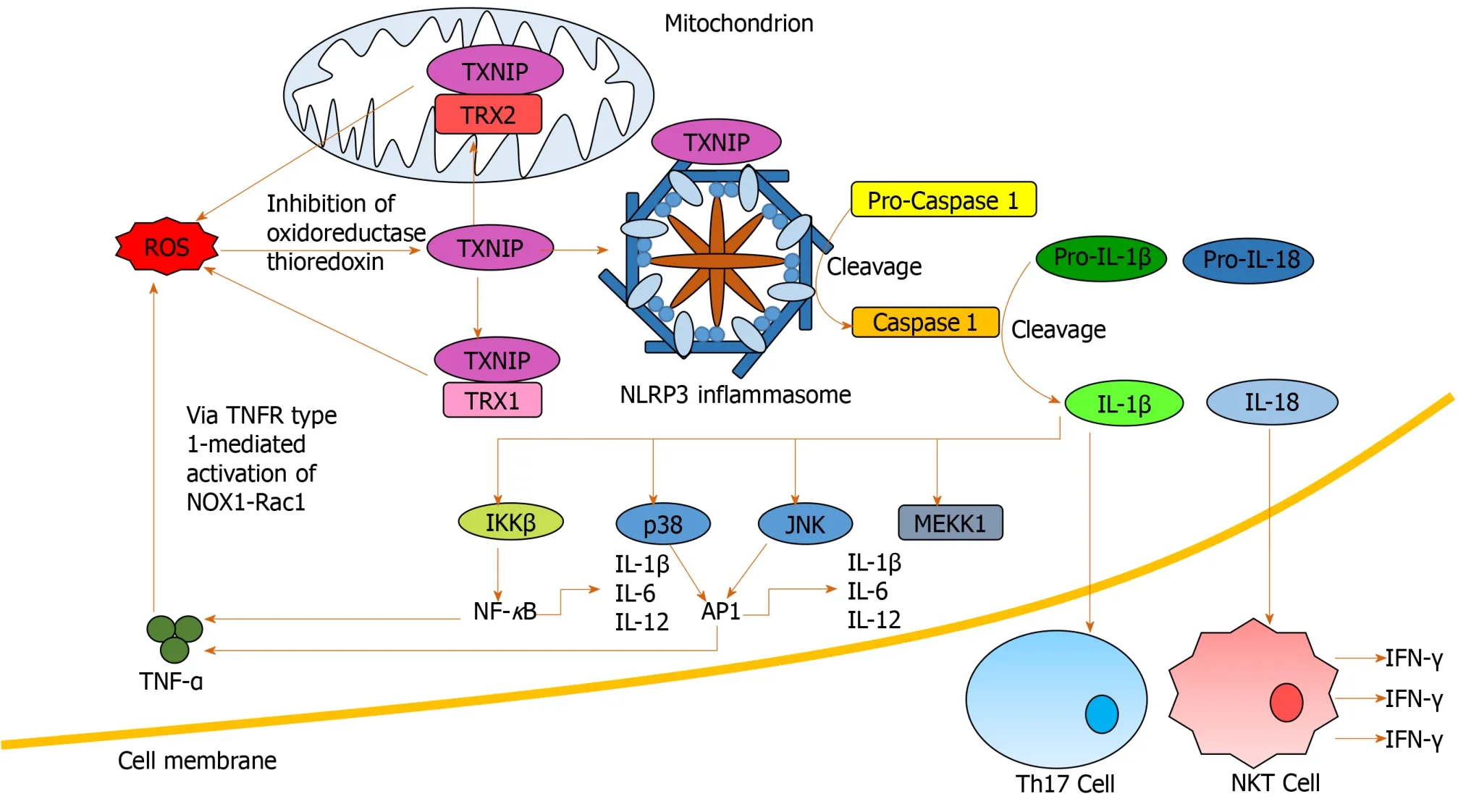
Figure 4 Nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 inflammasome activation and downstream signaling.
Reduced serum levels of trace elements have been confirmed in patients with liver disease, including ALD, and correlate with severity[107-110].Decreased zinc is associated with liver cirrhosis in alcohol dependent individuals[111]and reduced serum levels of zinc, copper and iron have been observed when compared with healthy controls[112].Zinc is a crucial trace element involved in multiple cellular and metabolic pathways[113]as well as acting as a cofactor for ALDH.Deficiency or abnormality in zinc function is implicated multiple pathologies, including liver disease (both acute and chronic)[114,115]and is associated with immune dysfunction evidenced by increased inflammation and aberrant immune cell activation[116].Zinc deficiency in endothelial cells results in increased oxidative stress and decreased inflammatory regulation which is corrected or partially ameliorated by zinc supplementation[117,118].In alcohol-fed mice, zinc deficiency worsens the balance between hepatic pro- and antioxidant enzymes[119]and is associated with accumulation of ROS in gut epithelial cells and disruption of tight junctions[120].Given zinc’s influence on antioxidant responses, gut integrity and immune function, a trial of zinc supplementation to improve clinical outcomes in patients with ALD cirrhosis is ongoing (NCT02072746).Preliminary reports suggest zinc supplementation is associated with a reduction in liver inflammation and improvement in immune function[121].
Antioxidant therapy may also have a benefit in the treatment of AH.An antioxidant cocktail (including zinc and selenium) in combination with steroids for the treatment of severe AH correlated with a significant reduction in serum biomarkers, improved short-term prognosis and reduced length of stay in hospital[122].However, a subsequent study of a complex regimen of N-acetylcysteine (NAC) followed by antioxidant therapy, alone or in conjunction with steroids, reduced renal injury but resulted in no survival benefit over 6 mo[121].Another clinical trial of steroids combined with NAC in AH showed reduced infection rate but not mortality at 6 mo[123].Antioxidants have also been shown to have a protective effect in patients with NAFLD by reducing serum levels of alanine transaminase (ALT) and spleen size, a finding that likely correlates with an improvement of fatty infiltration[122].These findings suggest that targeting or counterbalancing oxidative stress in ALD patients may improve patient outcomes.
EPIGENETICS
Lifestyle and environmental factors can modify gene expression without altering the DNA sequence, which gets transmitted to the next generation of cells after mitotic division, termed epigenetics[123].Epigenetic regulation includes both DNA and histone protein modifications as well as action through non-coding micro RNAs[123].DNA methylation is the most abundant epigenetic modification that directly affects the function of a gene in eukaryotes[124].Acetylation and deacetylation are modifications in histone proteins carried out by two enzyme families, histone deacetylases (HDACs) and histone acetyl transferase (HAT)[124].Histone modifying enzymes contribute to the activation or inactivation of transcription by catalyzing the unfolding or further compaction, respectively, of chromatin structure[124].
Excessive ROS is involved in epigenetic gene activation or silencing by changing DNA methylation levels[125].ROS production induces alterations in DNA methylation patterns and global histone acetylation, which then lead to aberrant gene expression, and may contribute to the process of carcinogenesis[124].The reduction of global histone acetylation in short term oxidative stress might be due to an immediate increase of class I/II HDAC activity by an unknown mechanism[126,127].Class III HDAC (Sirtuin NAD+-dependent family of protein deacetylases) has been hypothesized to be upregulated under oxidative stress because NAD+levels increase in the mitochondria under oxidative stress conditions but direct evidence is lacking[126].
Alcohol consumption increases gene-selective acetylation of histone H3 at lysine 9 (H3K9), levels of enzymes mediating histone acetylation, and results in a generalized increase in DNA methylation[126,127].These epigenetic-mediated effects of alcohol consumption regulate the inflammatory response, through key pro-inflammatory cytokines, such as TNF-α, which is silenced by H3K9 methylation and activated by H3K9 acetylation[128].In a macrophage cell line, alcohol treatment resulted in global increased histone H3 and H4 acetylation and specifically increased acetylation of proinflammatory gene histones[129].
Oxidative stress itself is an important regulator of epigenetic processes by inhibition of HDAC expression[130].This takes placeviaactivation of PI3Kδ, a signalling molecule controlling many inflammatory signalling pathways[131].Drugs that inhibit PI3Kδ (e.g., theophylline, nortriptyline and specific inhibitors) reduce oxidative stress inin vitroandin vivomodels of lung disease[132].In patients with AH, there isin vitroevidence that theophylline can enhance response to corticosteroid treatment, which may be mediated by its epigenetic effects[133].Targeting epigenetic regulation has recently been shown to have a beneficial effect in patients with AH; a novel sulphated oxysterol, DUR-928, was well tolerated and improved liver biochemistry in a small phase 2 clinical trial in AH[134].
Activation of the transcription factor Nrf2 is central to cellular defence against ROS[135].Its negative regulator, kelch-like ECM-associated protein 1 (Keap1), promotes proteasomal degradation of Nrf2.ROS decouples Nrf2 from Keap1, allowing it to translocate to the nucleus to bind to antioxidant response elements (AREs), initiating a range of antioxidant processes[135,136].Both Nrf2 and Keap1 expression are influenced by epigenetics with evidence of DNA hypermethylation in the Nrf2 promoter[135,136]and Keap1 promoter[137].Histone acetylation and deacetylation also modify AREdependent gene expression with Class 1 HDACs reducing Nrf2[138].Conversely, HDAC inhibitors restore Nrf2 expression and antioxidant responses.Targeting epigenetic regulation of Nrf2/Keap1 to ameliorate oxidative stress induced inhibition of antioxidant responses is an appealing strategy[138].However, much of this work has been performed in cancer cell lines and needs further investigation in the context of ALD.
IMPLICATIONS FOR THERAPY OF ALD
An improved understanding of the detailed mechanisms by which oxidative stress influences liver damage in patients with ALD may yield new targets for therapy.Current data from pre-clinical and clinical studies suggest potential new avenues for therapy of ALD.
MITOCHONDRIAL STRESS
Chronic alcohol consumption results in significant mitochondrial ROS generation leading to morphological and functional changes.Preventing ROS generation may ameliorate this process.Pre-clinical and early phase clinical studies have shown promise of this approach with SAMe.A systematic review and meta-analysis of 11 randomized controlled trials of SAMe treatment for chronic liver disease concluded that it improved liver biochemistry (bilirubin and AST) and had a good safety profile but did not affect mortality[139].Long term SAMe treatment in patients with ALD does not appear to be clinically effective with no reduction in adverse events or mortality in the two included studies performed in patients with ALD[140,141].However, short term treatment of the acute mitochondrial stress seen in AH may be a better strategy for the use of SAMe.A phase 2 clinical trial of SAMe with prednisolone for the treatment of severe AH demonstrated improved response rate measured by Lille score and a reduction in hepatorenal syndrome[54].However, there was no statistically significant difference in 28-d mortality.It may yet prove to be an effective adjunct to antiinflammatory therapy for AH.
UCPs are strongly associated with mitochondrial stress in ALD.Overexpression of UCP2 reduces apoptosis and oxidative stressin vitro[142].Hepatocellular downregulated mitochondrial carrier protein (HDMCP) expression induced uncoupling and reduced steatosis in an animal model of NAFLD[143].However, such an approach may promote hepatocyte necrosis and increase the risk of hepatocellular carcinoma[144].Further studies in this area are required to determine whether targeting UCPs would be a beneficial therapeutic strategy.
ANTIOXIDANT THERAPY
NAC, an antioxidant therapy that provides cysteine for glutathione synthesis, has been tested in patients with AH.Although initial trials did not demonstrate a survival benefit[145,146].a more recent study of NAC in combination with prednisolone, showed a reduction in infective events and 1-month mortality[147].Therefore, NAC has been suggested for the treatment of AH in clinical practice guidelines, with the caveat that a definitive randomized controlled trial is still required[148].
Deficiency of key trace elements is associated with oxidative stress, which is ameliorated by supplementation.Antioxidant therapy including zinc and other trace elements has shown clinical benefit in patients with AH[122].However, interpretation is hampered by use of a variety of antioxidants at differing concentrations and durations[145,146].A trial of long-term zinc supplementation in ALD patients has demonstrated improvements in short-term immune function[149]with long-term clinical outcomes due to be reported shortly.Improved understanding of the role of trace elements in ALD and the optimal formulation and duration of treatment is required.
EPIGENETIC REGULATION
Oxidative stress reduces HDAC expressionviaPI3Kδ activation resulting in increased expression of pro-inflammatory genes.Studies targeting HDACs have yet to be performed in patients with ALD.Althoughin vitrostudies suggest an antioxidant effect of HDAC inhibition with upregulation of Nrf2 expression[138], HDAC inhibitors approved for use in the treatment of cancer induce cell cycle arrest, apoptosis and oxidative stress in cancer cells which overexpress HDAC[150].The effect of HDAC inhibitors in the context of ALD requires carefulin vitroconfirmation before clinical translation.However, targeting PI3Kδ is a more appealing strategy with evidence from the respiratory field that specific inhibitors reduce oxidative stressin vitroandin vivo[132].
CONCLUSION
Alcohol is a major global healthcare and economic burden and is a growing cause of chronic liver disease.However, there are currently no effective therapies to treat ALD.Oxidative stress is involved in multiple aspects of ALD pathogenesis (Figure 5).Chronic alcohol consumption results in the saturation of the ADH pathway and increased CYP2E1-mediated alcohol metabolism.This leads to the generation of reactive species including MAA, HNE, lipid hydroperoxides, RNS and ROS, which cause hepatic damagevialipid and protein peroxidation, adduct formation and cellular hyper-regulation.Similar damage occurs in hepatic mitochondria with ROS inducing structural and functional damage.ROS cause oxidative damage through multiple mechanisms: Promoting cell deathviaprotein mediators, increasing and sustaining the upregulation of pro-inflammatory mediators, as well as inducing multiple epigenetic modifications.
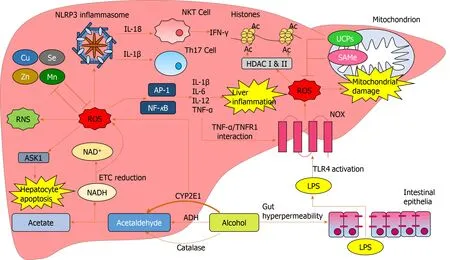
Figure 5 Reactive oxygen species-mediated oxidative damage in the liver.
 World Journal of Hepatology2020年7期
World Journal of Hepatology2020年7期
- World Journal of Hepatology的其它文章
- Is right lobe liver graft without main right hepatic vein suitable for living donor liver transplantation?
- Diagnosis and management of hepatic artery in-stent restenosis after liver transplantation by optical coherence tomography: A case report
- Effect of zinc treatment on clinical outcomes in patients with liver cirrhosis: A systematic review and meta-analysis
- Non-alcoholic steatohepatitis and the risk of myocardial infarction: A population-based national study
- Anti-inflammatory and anti-oxidant effects of aloe vera in rats with non-alcoholic steatohepatitis
- Ipragliflozin-induced improvement of liver steatosis in obese mice may involve sirtuin signaling