
97例糖原累积病的临床及病理特点分析
2021-01-11 11:46王丽苹何婷婷崔延飞王仲霞景婧王立福朱云孙永强许文涛余思邈桑秀秀田淼王睿林
肝脏 2020年12期
王丽苹 何婷婷 崔延飞 王仲霞 景婧 王立福 朱云 孙永强 许文涛 余思邈 桑秀秀 田淼 王睿林
糖原累积病(glycogen storage disease,GSD)是一类隐性遗传性糖原代谢紊乱性疾病,由于糖原代谢过程中某些酶的先天性缺乏致导致糖原分解或合成障碍,使糖原或异形糖原过多地累积在肝、肾、骨骼肌、心肌及中枢神经系统等组织中,从而出现肝脏肿大、肌张力降低或肌痉挛、低血糖、乳酸血症等临床表现的一组疾病。其中肝脏是主要的受累器官,故又称肝糖原累积病[1]。GSD临床较为罕见,本文对97例确诊为GSD患者进行回顾性研究,探讨GSD的临床及病理特点。
资料与方法
一、一般资料
收集解放军总医院第五医学中心2010年1月至2019年11月经肝组织病理诊断为GSD住院患者97例,男性75例,女性22例,年龄范围1~31岁,平均(4.7±5.9)岁,其中年龄为20岁以上患者仅6例。
二、方法
(一)观察指标 患者的性别、年龄、临床症状、体征、总胆红素(TBil)、直接胆红素(DBil)、血清丙氨酸氨基转移酶(ALT)、天冬氨酸氨基转移酶(AST)、碱性磷酸酶(ALP)、γ-谷氨酰基转移酶(GGT)、空腹血糖(GLU)、总胆固醇(TC)、三酰甘油(TG)、低密度脂蛋白胆固醇(LDL-C)、高密度指蛋白胆固醇(HDL-C)、尿素氮(UREA)、肌酐(CRE)、尿酸(UA)、乳酸(LAC)及病理特点。
(二)组织学 超声引导下采用快速穿刺法取肝组织。标本长1~2 cm,镜下包括至少6个汇管区。常规石蜡包埋、切片、脱蜡、HE染色、Masson染色;periodic acid schiff染色法(PAS法)观察肝组织内糖原沉积。
结 果
一、临床特点
97例GSD患者均存在不同程度的肝大和肝功能异常,伴不同程度的低血糖 73例,3例出现低血糖惊厥;低体重24例,生长迟缓11例,消瘦8例;腹部膨隆45例,脾大10例,发热8例;贫血27例,其中轻度贫血24例,中度贫血3例,未见重度及极重度贫血;心肌损伤2例,IgA肾病1例。97例GSD患者中有12例存在GSD家族史。
二、实验室指标
TBil(12.11±15.54)μmol/L,DBil(4.89±7.51)μmol/L,ALT(262.54±221.92)U/L,AST(432.16±394.80)U/L,ALP(272.63±88.44)U/L,GGT(183.10±405.63)U/L,GLU(3.19±1.26)mmol/L,TC(4.76±1.62)mmol/L,TG(3.19±2.74)mmol/L,LDL-C(3.43±1.06)mmol/L,HDL-C(0.85±0.27)mmol/L,UREA(4.18±1.22)mmol/L,CRE(37.55±9.72)μmol/L,UA(324.04±98.53)μmol/L,LAC(3.85±3.47)mmol/L。
三、病理表现
97例患者均经肝组织病理学检查确诊为GSD。肝穿病理组织光镜下主要病理改变:部分区域小叶结构紊乱,肝细胞弥漫性肿胀和或增大,部分胞质空化、核小居中,空泡状核易见,胞膜清晰,肝细胞呈植物细胞样,少数点灶状坏死;少量混合性炎细胞浸润,窦周纤维化可见;汇管区扩大,少量炎细胞浸润,细纤维隔形成,未见明确界面炎。肝脏病理PAS染色均为阳性。炎症程度G0 17例, G1 74例,G2 6例,纤维化程度S1 22例,S2 31例,S3 26例,S4 18例,其中17例可见肝细胞不同程度的脂肪变性(图1),2例多发性肝腺瘤(图2),1例(1.0%)合并肝癌(图3)。
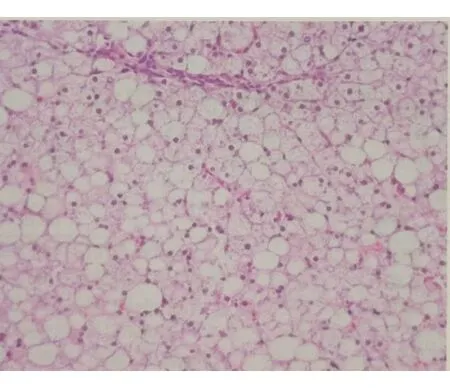
图1 GSD重度脂肪变性(HE染色×100)

图2 GSD多发性肝腺瘤(HE染色×100)
四、治疗
97例患者采取高碳水化合物、足量蛋白质、低脂饮食,并少量多餐。禁蔗糖、限乳糖和果糖,控制甜食及水量的入量。对乳酸高的患者建议服用免乳糖奶粉,尿酸高的患者限制饮食中嘌呤的含量。并给予生玉米淀粉口服治疗。嘱用凉白开冲生玉米淀粉(水∶淀粉=2∶1),间隔6 h两餐中间服用,每次1.6 g/kg。并给予还原型谷胱甘肽、多烯磷脂酰胆碱等保肝、降酶及对症支持治疗。大部分患者经过治疗后腹部膨隆明显改善,肝功能基本恢复正常,肝脏较前缩小,体质量增长。
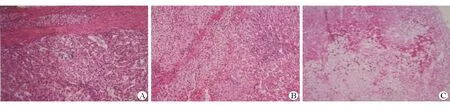
A:肝细胞癌区域(HE染色×200);B:腺瘤区域(HE染色×100);C:糖原累积性肝硬化Ⅰ型[PAS染色(灶性+)]
讨 论
GSD是一类由于基因缺陷导致在糖原合成或水解过程中酶缺乏或活性降低,引起机体能量代谢障碍和糖原在组织中过多沉积的遗传性糖代谢障碍疾病[2]。根据缺陷酶的不同及发现的年代顺序糖原累积病被分为十几个类型[3],肝糖原累积病占9型,其中Ⅰ、Ⅲ、Ⅳ型肝脏损伤最严重[4]。肝糖原累积病是儿童期引起肝功能损伤的常见疾病之一[5、6],肝糖原累积病患儿起病时的年龄、病情进展的速度、其他器官的累及程度、临床表现均有较大差异。
本研究发现,GSD以儿童期为主,男性多于女性;17例患者在肝大、肝功能异常基础上同时伴发低血糖、高脂血症、高尿酸、高乳酸;其肝脏组织病理表现为肝细胞染色较淡,浆膜明显,胞质内充满糖原且含有中等或大的脂肪滴,细胞核内糖原累积、明显肝脂肪变性但无明显纤维化改变,符合GSDⅠ型表现[4、7]。GSDⅠ型是由于葡萄糖-6-磷酸酶缺乏所致,是婴幼儿中常见的一种GSD。GSDⅠ型分为Ⅰa、Ⅰb、Ⅰc、Ⅰd 4种亚型。GSDⅠa型又称Von Gierke病[8],该亚型约占80%,主要表现为肝、肾肿大,生长发育迟缓及高脂血症,幼儿患者可因严重低血糖而夭折,不伴有脾脏肿大。本研究中患者多数有空腹低血糖,但无明显临床症状,有3例出现低血糖惊厥。黄巧燕等[9]报道,18例以意识障或惊厥为首发症状的儿童低血糖症临床特点和误诊分析中2例考虑为糖原累积症Ⅰ型。王小军等[10]报道难治性小儿低血糖症20例临床分析中3例诊断糖原累积症。针对这一现象可能是由于葡萄糖-6-磷酸酶的活性减低或缺乏,影响肝糖原转化为葡萄糖,糖原蓄积于肝细胞内,造成肝大、空腹低血糖,从而促使脂肪代谢增加,造成血脂、血乳酸、血尿酸增高等代谢紊乱[11]。在低血糖情况下高乳糖血症能够为大脑提供另一种能量来源[1]
肝脏腺瘤是GSDⅠa型最常见的并发症,1955年首次报道GSD患儿容易合并发生肝脏腺瘤[12]。本研究中有2例多发性肝腺瘤患者均为12岁,1例合并肝癌患者22岁。与唐晓艳等[13]报道糖原累积症Ⅰa型致青春期左右易发生肝脏腺瘤,部分癌变相符。肝脏腺瘤在基因方面研究,有提出可能由于染色体基因异常,导致肝脏腺瘤更易发生,且更可能发生癌变[14、15]。肝脏多发腺瘤的GSDⅠ型患者的最佳临床治疗方案,目前仍然存在争议[16]。夏晓刚等[17]曾报道手术治疗合并肝脏多发腺瘤的Ⅰa型糖原累积症1例,对较大肿物针对性地切除、对小的腺瘤行射频消融的治疗方案,术后再根据病理进一步制定治疗方案。
GSD Ⅲ型是糖原脱支酶(GDE)活性缺失,使葡萄糖分解障碍,大量异常糖链蓄积于肝脏或肌肉中。主要表现为肝肿大,生长发育迟缓,常并发肝纤维化、心肌病。该型分为4种亚型,Ⅲa型占Ⅲ型的85%,多表现为肝肿大,低血糖,生长迟缓,心肌肥大[18]。本研究中有24例低体重、11例生长迟缓、8例消瘦、2例心肌损伤。肝组织病理:纤维化程度S1 22例,S2 31例。GSDⅠ型和GSDⅢ型在肝组织病理特点方面有两点相鉴别:①肝组织明显纤维化,可有纤维间隔;②肝细胞缺乏脂肪变性,很少有明显肝硬化。因肝组织纤维化明显,最终会导致肝脾肿大等门静脉高压表现。本研究中的97例患者中有36例患者临床特点较符合GSDⅢ型。
GSDⅣ型是由于糖原合成途径中糖原分支酶(GBE)缺乏所致,约占所有GSD的0.3%[19]。该型可分为3种亚型:婴幼儿型、青年型和成人型。临床表现为出生时正常,随后出现生长发育迟缓及肝脾肿大,进展至肝硬化,最后由于门脉高压、腹水、食管静脉曲张、肝功能衰竭,一般于出生后3~5岁死亡。其肝脏组织病理特点为肝脏呈小结节性肝硬化伴宽纤维束围绕或插入肝小叶。门脉区胆管轻度增生,肝小叶周边细胞内可发现嗜酸性或无色包涵体沉积在细胞质,把肝细胞核推向一侧[20]。本研究中患者年龄小于2岁共47例,其中44例患者空腹血糖正常,无高脂血症;就诊时尚无失代偿期临床表现,肝组织病理中26例出现肝硬化趋势(S3),18例典型肝硬化(S4)。因此,本研究中有44例患者高度怀疑为GSD Ⅳ型。
肝组织酶活性检测是明确肝糖原累积病分型的“金标准”[1]。根据病史、体征及血生化检测可作出初步临床诊断,随着技术的发展,通过对突变基因进行全基因测序的无创分子遗传学检测已成为诊断GSD的首选方法,准确,快速,同时避免肝活检对机体带来的损害。基因测序分析诊断GSD在临床上正逐渐代替肝活组织检查。20世纪80年代开始的生玉米淀粉治疗可有效纠正低血糖,严格遵守饮食疗法能使GSD患者生长和青春期发育接近正常。在治疗上尚无成熟的方案,多采用饮食疗法,对于药物治疗失败可能需要器官移植,肝移植是肝脏代谢疾病的终极疗法。
猜你喜欢
传染病信息(2022年6期)2023-01-12
广西糖业(2022年5期)2022-11-24
中国临床医学影像杂志(2022年2期)2022-05-25
中国典型病例大全(2022年13期)2022-05-10
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2022年1期)2022-02-28
天津医科大学学报(2021年2期)2021-03-29
中国临床医学影像杂志(2019年5期)2019-08-27
实用肿瘤学杂志(2019年5期)2019-02-10
成都体育学院学报(2017年1期)2017-02-21
