
SR1001通过干扰PI3K/AKT/mTOR通路磷酸化抑制艰难梭菌感染*
2021-01-06 03:15王浦陈烨
胃肠病学 2020年7期
王 浦 陈 烨
南方医科大学南方医院消化内科(510515)
背景:艰难梭菌感染(CDI)是最常见的机会致病菌感染,抗菌药物是一线治疗用药,但存在治疗效果不佳、感染复发、再燃等情况,有必要开发新型的抗感染药物。目的:探讨SR1001对CDI的干预作用及其潜在机制,探寻潜在的干预靶标。方法:培养艰难梭菌(C.difficile),将结肠癌HT-29细胞分为对照组、CDI组(感染C.difficile)和SR1001治疗组(感染C.difficile+SR1001干预治疗),观察HT-29细胞形态改变情况,CCK-8法检测细胞增殖情况,蛋白质印迹法检测PI3K/AKT/mTOR信号通路表达情况,ELISA法检测细胞上清液中TcdB含量。结果:与对照组相比,CDI组细胞透亮变圆,凋亡明显;细胞增殖受抑制情况随时间延长而加重,且细胞增殖能力明显低于对照组(P<0.05),PI3K/AKT/mTOR通路磷酸化蛋白表达明显升高,上清液中TcdB含量明显增高(P<0.05)。给予SR1001治疗后,细胞趋于正常“铺路石”样排列,凋亡改善,细胞增殖能力明显高于CDI组,PI3K/AKT/mTOR通路磷酸化蛋白表达明显降低,上清液中TcdB含量明显减少。结论:SR1001可通过干扰PI3K/AKT/mTOR信号通路磷酸化,逆转C.difficile对结肠癌细胞的生长抑制作用。
艰难梭菌(C.difficile)是一种革兰阳性厌氧芽孢杆菌,艰难梭菌感染(Clostridiumdifficileinfection, CDI)是目前最常见的医院获得性感染之一[1]。C.difficile定植于肠道,当宿主肠道菌群失衡时,C.difficile大量生长,释放毒素TcdB,造成细胞骨架破坏,凋亡坏死,破坏上皮屏障,引起CDI的发生。轻症感染仅引起腹泻,重症感染可发展为伪膜性肠炎,甚至中毒性巨结肠、肠穿孔等并发症。
辅助性T细胞17(Th17细胞)参与多种感染性疾病和炎性疾病[2-3]。有研究表明,Th17细胞通过分泌IL-17诱发炎症反应,促进CDI的发生[4-5]。Yu等[6]通过检测CDI中的IFNs,发现重症CDI患者Th1细胞转变为Th17细胞。维甲酸相关孤核受体(RORs)参与Th17炎性细胞IL-17分泌等功能的调控[7]。SR1001可非特异性拮抗RORα与RORγt活性,抑制IL-17的产生,从而改善疾病的症状,减缓疾病的发生、发展。本研究通过建立C.difficile细胞感染模型并给予SR1001药物干预,旨在探讨SR1001对CDI的治疗作用及其机制。
材料与方法
一、细胞和主要试剂
人结肠癌HT-29细胞由南方医科大学南方医院消化科保种培养传代,C.difficile菌株购自ATCC公司,DMEM培养基和胰酶购自Hyclone公司,胎牛血清购自Gibco公司,CCFA和BHI购自北京索莱宝科技有限公司,SR1001购自MedChemExpress公司,PI3K、AKT、mTOR、p-AKT、p-mTOR购于Abcam,p-PI3K购于北京博奥森生物技术有限公司,CCK-8检测试剂盒购于碧云天生物技术有限公司,人艰难梭菌毒素B(TcdB)ELISA试剂盒购于江苏酶标生物科技有限公司。
二、研究方法
1.C.difficile培养:将C.difficile菌株接种于CCFA培养皿中。37 ℃厌氧培养48 h后,接种于BHI液体培养24 h,采用紫外分光光度计测量波长600 nm处的吸光度值(A值),将菌悬液浓度调整至1×108CFU/mL,2 000 r/min 4 ℃离心10 min,取上清液,用于后续细胞实验。
2.细胞培养和分组:结肠癌HT-29细胞中加入含10%胎牛血清的DMEM培养基,置于5% CO2、37 ℃的恒温箱中培养,传代培养。使用0.25%胰蛋白酶消化细胞,制备细胞悬液并接种于96孔板,待细胞生长融合至80%时,将细胞分为对照组、CDI组和SR1001治疗组,对照组不作任何处理,CDI组细胞加入1×108CFU/mLC.difficile培养上清液,SR1001治疗组加入1×108CFU/mLC.difficile培养上清液+5 μmol/L SR1001,孵育48 h。
3.CCK-8法检测细胞增殖:取各组对数生长期细胞,接种于96孔板上,每孔约2 000个细胞。分别于培养0、12、24、48 h时加入10 μL CCK-8溶液孵育2 h,上酶标仪检测450 nm波长处的A值,绘制生长曲线。
4.蛋白质印迹法:将各组HT-29细胞铺于6孔板中,RIPA裂解法提取蛋白,BCA蛋白定量后,经电泳、转膜、封闭、孵育抗体(PI3K、AKT、mTOR、p-PI3K、p-AKT、p-mTOR工作浓度均为1∶1 000;GAPDH工作浓度为1∶20 000),ECL发光显色,检测PI3K/AKT/mTOR通路磷酸化蛋白表达情况。
5.ELISA法:根据试剂盒说明书,取各组孵育48 h后的细胞培养上清液,检测TcdB含量。
三、统计学分析
结 果
一、SR1001减轻C.difficile对HT-29细胞的毒性作用
与对照组(图1A)相比,CDI组细胞变圆透亮,凋亡明显,失去正常细胞生长形态(图1B)。SR1001治疗组细胞部分恢复生长形态,呈“铺路石”样排列(图1C)。

A:对照组;B:CDI组;C:SR1001治疗组
二、SR1001可拮抗C.difficile的抑制作用
CCK-8法结果显示,CDI组细胞增殖受到明显抑制,并随时间延长而加重(P=0.00),48 h时细胞增殖能力明显低于对照组(P<0.05),而给予SR1001治疗后,细胞增殖能力有所改善(P<0.05;图2、表1)。证实SR1001可通过改善细胞增殖能力来发挥抑制C.difficile毒性的作用。
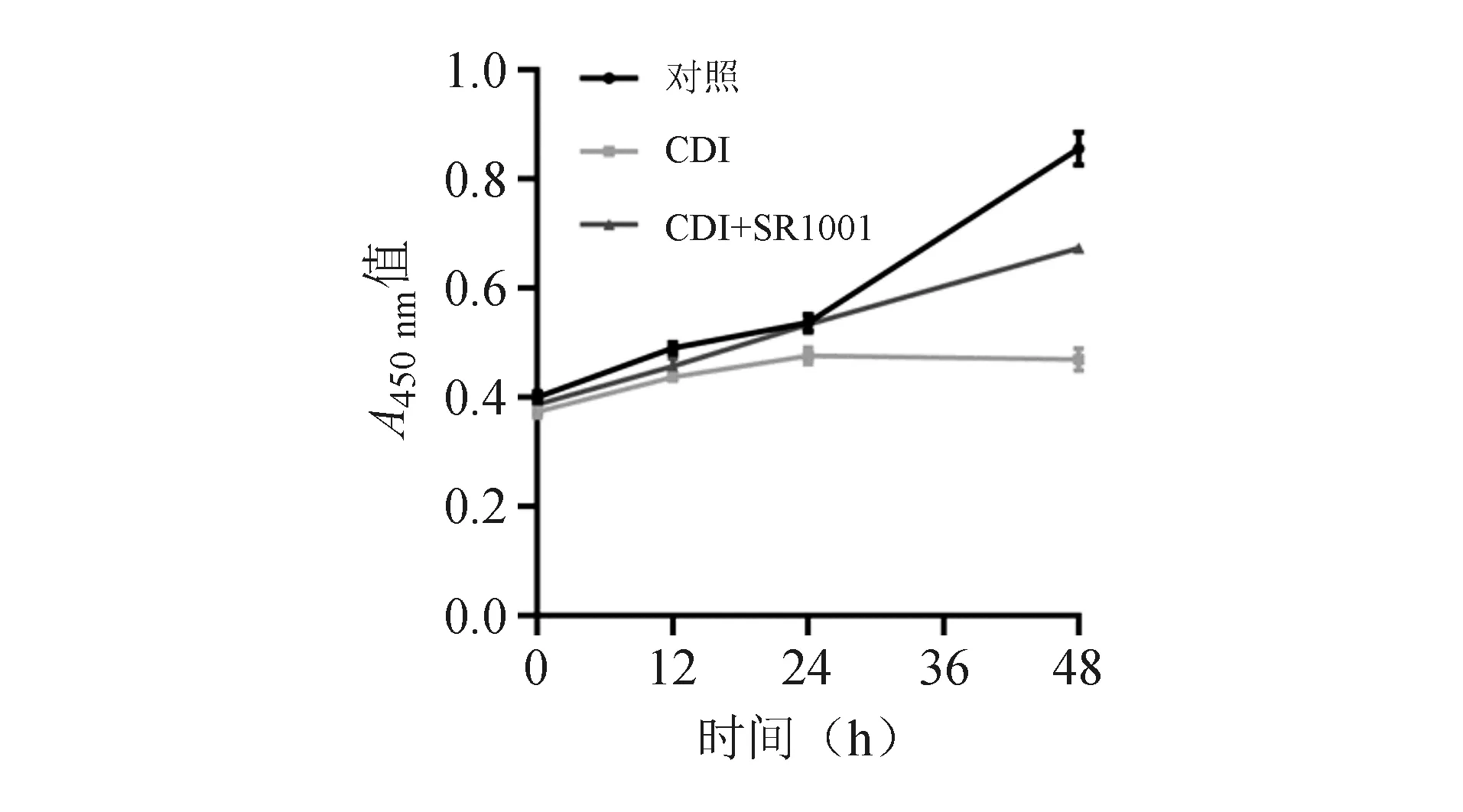
图2 CCK-8实验测定不同时间点HT-29细胞的增殖能力
三、SR1001可降低C.difficile感染HT-29细胞后PI3K/AKT/mTOR通路磷酸化蛋白表达
蛋白质印迹法结果显示,C.difficile感染可使p-PI3K、p-AKT、p-mTOR表达较对照组明显升高,经SR1001治疗后,p-PI3K、p-AKT、p-mTOR蛋白表达明显降低(图3、表1),而三组PI3K、AKT、mTOR总蛋白含量无明显差异。提示SR1001可通过调控PI3K/AKT/mTOR通路改善C.difficile感染情况。

1 Da=0.992 1 u
表1 SR1001对细胞增殖、PI3K/AKT/mTOR通路磷酸化蛋白表达以及C.difficile定植的作用
四、SR1001可抑制C.difficile定植
ELISA法结果显示,与对照组相比,CDI组细胞上清液中TcdB含量明显升高,给予SR1001治疗后,上清液中TcdB含量明显降低(图4、表1)。说明SR1001可抑制C.difficile在细胞中的定植。
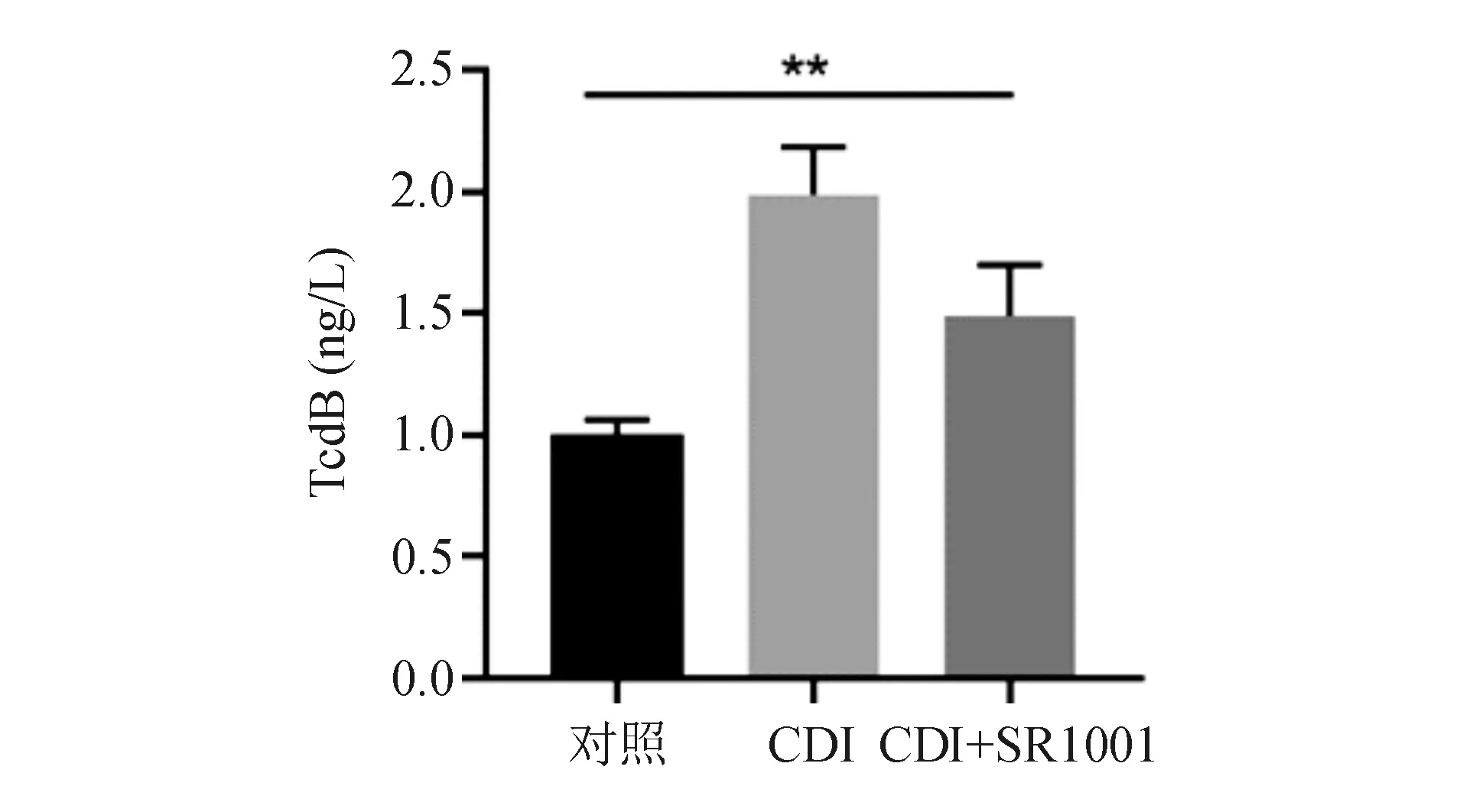
图4 ELISA法测定HT-29细胞培养上清液中TcdB含量
讨 论
近年来,CDI居高不下的感染率越来越引起关注[8]。C.difficile毒素TcdB作用于肠上皮细胞,破坏细胞骨架,诱导细胞坏死凋亡,破坏上皮完整性[9],从而诱导多种病变。多种因素参与CDI的发生、进展,炎症反应是CDI发生过程中的重要因素,Th17细胞是促炎细胞的亚群之一,可促进炎症反应,分泌多种促炎因子[10]。IL-17是Th17细胞分泌的重要的炎症因子,IL-17失衡可引起过度反应,从而导致肠道组织破坏,RORs作为特异性转录因子可调控IL-17分泌。目前,多种研究表明,RORs特异性逆激动剂可有效控制与Th17细胞相关的自身免疫病[7,11-14]。SR1001是一种高选择性和特异性RORs逆激动剂,可抑制Th17细胞分化和功能[7]。本研究中,以SR1001干预C.difficile感染细胞,可明显改善细胞毒性作用,逆转C.difficile对细胞增殖的抑制作用,并可抑制C.difficile定植。
Th17细胞产生IL-17作为Th亚群参与上皮细胞和中性粒细胞介导的多重免疫反应,抵抗外来微生物,并参与各种自身免疫病的发展。Th17细胞的分化受细胞内一系列信号级联和复杂的转录因子网络的控制。最近研究表明,PI3K/AKT/mTOR通路参与细胞代谢、增殖、分化等诸多过程,该通路通过调节STAT3磷酸化、调控HIF-1α表达以及激活RORγt核移位、下调Gfi1等多种机制正向调节Th17细胞的分化,是Th17细胞形成的重要经典途径[15]。本研究结果发现C.difficile感染细胞后,p-PI3K、p-AKT、p-mTOR表达较对照组明显升高,而SR1001干预治疗后,p-PI3K、p-AKT、p-mTOR蛋白表达较CDI组均有所下降,说明SR1001可通过干预PI3K/AKT/mTOR通路抑制C.difficile感染。
综上所述,本研究通过建立C.difficile感染的体外模型,并给予SR1001干预治疗,揭示SR1001可通过下调p-PI3K、p-AKT、p-mTOR表达来抑制PI3K/AKT/mTOR通路,从而缓解C.difficile毒素作用,促进细胞增殖,同时可抑制C.difficile定植,为进一步研究CDI的免疫机制,探寻CDI干预靶标,以及CDI的防治提供了一定的参考和依据。
猜你喜欢
生殖医学杂志(2022年10期)2022-10-19
当代水产(2022年1期)2022-04-26
当代水产(2022年2期)2022-04-26
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
中国饲料(2021年17期)2021-11-02
当代水产(2021年2期)2021-03-29
中小学德育(2020年11期)2020-03-18
分析化学(2017年12期)2017-12-25
教育界·上旬(2016年12期)2017-05-25
