
The Role of Environmental Stress in Determining Gut Microbiome:Case Study of Two Sympatric Toad-headed Lizards
2020-12-30 06:59:28YueQIWeiZHAOYangyangZHAOXiaoningWANGandChenkaiNIU
Asian Herpetological Research 2020年4期
Yue QI,Wei ZHAO*,,Yangyang ZHAO,Xiaoning WANG and Chenkai NIU
Gansu Key Laboratory of Biomonitoring and Bioremediation for Environmental Pollution,School of Life Sciences,Lanzhou University,Lanzhou 730000,Gansu,China
Abstract The gut microbiota has gained attention because of its importance in facilitating host survival and evolution.However,it is unclear whether gut microbial communities are determined by the host (heritable factor) or environment (environmental factor).In this study,we investigated the gut microbial communities and potential functional signatures of two sympatric species distributed along an elevation gradient,the toadheaded lizards Phrynocephalus axillaris and P.forsythii.Our results indicated that at high elevations,the gut microbial communities of P.axillaris and P.forsythii did not significantly differ,and the phylogenetic relationships of gut microbial communities contradicted their hosts.At low altitudes,the two lizards could be distinguished based on their significantly different gut microbial communities.Compared to low-altitude populations,Kyoto Encyclopedia of Genes and Genomes (KEGG)pathway analysis showed that at higher altitudes,energy metabolism,such as carbohydrate,lipid,and amino acids metabolism were higher in both lizards.While a larger number of pathogenic bacteria were found in the lowaltitude population of P.forsythii.This suggests that the convergence of gut microbiota of two lizards at highaltitude stem from environmental factors,as they were exposed to the same environmental stress,whereas the divergence at low-altitude stemmed from heritable factors,as they were exposed to different environmental stresses.These results provide a new perspective regarding whether heritable or environmental factors dominate the gut microbiota during exposure to environmental stress.
Keywords gut microbiota,environmental factor,environmental stress,heritable factor,toad-headed lizard
1.Introduction
Microbes in a host are increasingly recognized as having fundamental roles in animal evolution (Wuet al.,2009).An individual is not necessarily a single organism,but part of a co-evolved community that includes the host and its microbes(Mcfallet al.,2012; Bordenstein and Theis,2015).In animals,the most numerous microbes are those in the gut,which are likely comprised a core microbiota and flexible microbial pool (Shapira,2016).As the 'second genome' of the host(Spanogiannopouloset al.,2016),the gut microbiota can provide a series of functions to their host which promote evolution,attracting the attention of biologists.
Two factors are typically considered to determine gut microbial communities:heritable and environmental factors(Moelleret al.,2013; Suzukiet al.,2019).The core microbiota,which may be important for basic functions in the host,is putatively shaped by host genetics (Roeselerset al.,2011).Further,the mutual relationship between the gut microbiota and their host facilitates vertical transmission of the gut microbiota(Clarket al.,2000; Colston,2017) and may cause phylogenetic congruence (Brucker and Bordenstein,2011).While a flexible microbial pool may facilitate adaptation of the host to a local niche,shaped by environmental horizontal transmission(Carmodyet al.,2015; Songet al.,2019).A previous study showed that the gut microbiota of genetically unrelated individuals who share the same field were remarkably similar (Rothschildet al.,2018).Further,by allowing gut microbiota replacement from the environment,the host and gut microbiota may show phylogenetic non-congruence (Kikuchiet al.,2012).However,the dominant factor determining the gut microbial community remains unclear (Bensonet al.,2010; Bruckeret al.,2011; Bergetal.,2016; Rothschildet al.,2018).
The two toad-headed lizards,Phrynocephalus axillarisandP.forsythii,which are sympatric at two different altitudes,can explore the dominating factor of determine the gut microbial community.We propose two alternative hypotheses:(1)heritable factors have a dominant contribution.If true,the compositions of the gut microbial communities will show interspecific variation in the same area but be intraspecific conservative.(2) Environmental factors make a dominant contribution,in which the gut microbial communities of these two species would converge in the same environment and show phylogenetic non-congruence.This research provides insight into the factors affecting gut microbial communities.
2.Materials and Methods
2.1.Study area and samplingPhrynocephalus forsythiiandP.axillariswere found to coexist at both high (Cele) and low(Korla) altitudes in the Tarim Basin (Figure 1).Korla is located in the southwest corner of the Tarim Basin and has a dry climate.According to the National Climatic Data Center(http://data.cma.cn/),the annual average temperature of the Korla is 12.0 °C and the average annual rainfall is 5.0 cm.While the elevation of the Cele sampling point is 2258; the oxygen levels are relatively low and the annual average temperature and annual precipitation are 10.2 °C and 5.53 cm,respectively.Six adult males were collected from each population of the two species.Since our aim was to compare the differences of gut microbiota between two populations,no negative controls were set.The geographical location of the sampling sites was determined using a Garmin Oregon E20 handheld GPS unit(Garmin,Olathe,KS,USA).
Animals were treated in accordance with the guidelines of Ethics Committee of the School of Life Sciences,Lanzhou University,which approved this study.
2.2.DNA extraction and sequencingThe full intestinal tract was dissected and collected into tubes under sterile conditions.Total genomic DNA was extracted using a Fecal DNA Extraction Kit from Sangon (DP328,Shanghai,China) and was stored at -20 °C until further analysis.The V3 and V4 regions of the bacterial 16S rRNA gene was amplified from the total extracted DNA using primers 341F and 806R,respectively.The amplification program comprised one cycle of 98 °C for 1 min,followed by 30 cycles of 98 °C for 10 s,annealing at 50 °C for 30 s,and elongation at 72 °C for 60 s; finally,the PCR system was held at 72 °C for 5 min.PCR products were separated by agarose gel electrophoresis and purified using a GeneJET Gel Extraction Kit (Thermo Scientific,Waltham,MA,USA).

Figure 1 Sampling sites of P.forsythii and P.axillaris.The altitudes of two sample sites were 895 m (Korla) and 2126 m (Cele),respectively.The elevation gradient is represented by graduated colors from red to blue (low to high).The map was downloaded from the National Fundamental Geographic Information System (http://nfgis.gsdi.gov.cn/).
The DNA libraries were constructed using a TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina,San Diego,CA,USA) following the manufacturer’s recommendations,with index codes were added.The components of the libraries were then sequenced on an Illumina Nova platform and paired-End sequencing was performed using sequencing strategy PE250 with the fragment size of read length,450-550 bp.Raw reads obtained by sequencing have been deposited to SRA(PRJNA623140).
2.3.Sequence assembly and taxonomic identificationMicrobial sequences were analyzed using QIIME v1.9.1(Caporasoet al.,2010).Raw reads obtained from sequencing were spliced to obtain raw tags,then clean tags were obtained after filtering low-quality and short-length raw tags; effective tags were used after filter the chimeric sequences for subsequent analysis.Next,the sequences were clustered into operational taxonomic units (OTUs) byde novoOTU picking at a 97%level of sequence similarity (Yanet al.,2015).The taxonomic assignments for each representative sequence were obtained using the GreenGenes v2.2 database (Wanget al.,2007).OTU abundance information was normalized using a standard sequence number corresponding to the sample with the lowest number of sequences.
2.4.Microbial community relationships among host speciesThe unweighted UniFrac distance between microbial communities of all individuals from high or low elevations were calculated using the Jaccard coefficient and were represented using an unweighted pair group method with arithmetic mean (UPGMA) clustering tree.The pairwise comparisons of community composition were calculated by computing unweighted UniFrac distances; while were visualized by ordination using principal coordinates analysis with the visualization software EMPeror (Vazquezet al.,2013).To identify differences in the microbial communities between the four groups,analysis of similarities was performed based on the unweighted UniFrac distances matrices by vegan package in R software (Clarke,1993).
2.5.Differences in microbial taxa abundance and microbial community functionsFour groups were compared:two intraspecific comparisons at different altitudes and two interspecific comparison at the same altitude.The abundance of the microbial taxa was expressed as a percentage of the total 16S rRNA gene sequences,and differences between groups were compared.A two-sided Welch’st-test was used to identify significant differences in microbial taxa.Statistical analyzes was performed using STAMP software (Parkset al.,2014).

Figure 2 Relative abundances of microbial phyla of P.axillaris and P.forsythii represented as bar plots at the phylum level (left) and genus level (right).Only the top ten phyla and top 30 genera are shown in the histogram,and the other taxa are combined.A and F indicate P.axillaris and P.forsythii,and H and L indicate high and low elevations,respectively.AL:P.axillaris from low altitude; AH:P.axillaris from high altitude; FL:P.forsythii from low altitude; FH:P.forsythii from high altitude.
To predict each metagenome based on the 16S rRNA amplicon sequences,we used the tool Phylogenetic Investigation of Communities by Reconstruction of Unobserved States(Langilleet al.,2013).The OTU data were rarefied and normalized according to predicted 16S rRNA gene copy numbers,and the metagenomes were predicted using the precalculated Kyoto Encyclopedia of Genes and Genomes(KEGG) orthologs database (https://www.genome.jp/kegg/).Predicted metagenomes were collapsed into a specified level in a hierarchy using the KEGG pathway metadata and the relative abundance differences in the KEGG pathways of the four groups were compared by two-sided Welch’st-test (Parkset al.,2014).
3.Results
In total,1 749 891 raw reads were obtained from sequencing.Subsequently,1 701 632 clean tags were filtered from 1 730 109 raw tags obtained from 24 intestinal content samples.After filter the chimeric sequences,1 539 919 effective tags were used for subsequent analysis.Based on the rarefaction curves of the observed species (Figure S1),four individuals with large deviations from the overall level were deleted (AH01,AH02,FH03,FL06).At a 97% similarity level,4480 OTUs were obtained from 20 intestinal content samples.All OTUs were classified into 40 phyla,whereas 1861 (41.54%) OTUs were annotated into 683 genera.At the phylum level,the three dominant phyla detected in both groups wereProteobacteria,Firmicutes,andBacteroidetes.At the genus level,Bacteroideswas the predominant bacterial genus (Figure 2).
3.1.Microbial community composition and structureThe results of UPGMA clustering revealed that the bacterial community membership of all samples from high elevations were not distinguishable betweenP.axillarisandP.forsythii(Figure 3A).At low elevations,the relationships of the gut microbial communities of all individuals reflected the phylogeny of their host (Figure 3B).
Principal coordinates analysis revealed that the gut microbiota of the two lizards did not significantly differ at high altitudes (R=0.043,P=0.567),whereas large differences were observed at low altitudes (R=0.565,P< 0.001).In the intraspecific comparison group,the difference in the gut microbial communities inP.axillariswere not significant along the altitude gradient,whereas the differences in the gut microbial communities inP.forsythiiwere significant (R=0.476,P=0.016) (Figure 3C and Table 1).
3.2.Differentially abundant microbial taxaIn intraspecific comparison,the numbers of Erysipelotrichia,Erysipelotrichales,and Erysipelotrichaceae were significantly higher in the low attitude population ofP.axillaris,whereas the order Rickettsiales,family Anaplasmataceae,and genusWolbachiawere significantly higher in the low attitude population ofP.forsythii.No differences in the gut microbiota were detected in the interspecific comparison group at highaltitude (Figure 4 and Table 1).In the interspecific comparison group at low-altitude,some microbial taxa with significant differences in abundance were also detected in the gut microbiota of these two species.In the gut microbiota ofP.axillaris,the abundance of order Desulfovibrionales,family Desulfovibrionaceae,and generaBilophila,Intestinimonas,Oscillibacter,andTyzzerellawas significantly higher thanP.forsythii.In the gut microbiota ofP.forsythii,the abundance of order Entomoplasmatales,Rickettsiales and Rhizobiales,family Anaplasmataceae,and genusWolbachiawas significantly higher thanP.axillaris(Figure 4 and Table 1).
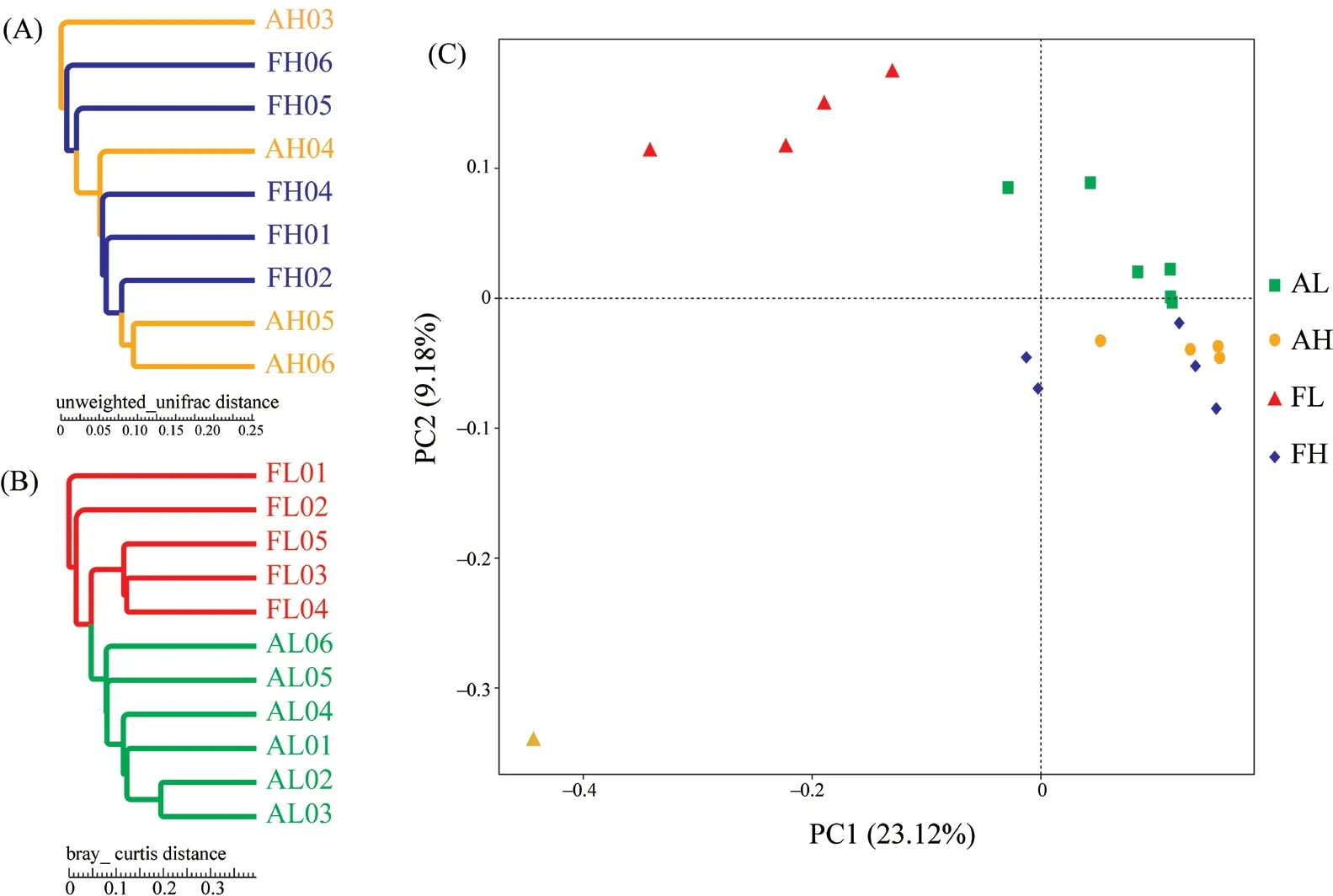
Figure 3 Comparison of gut microbial communities in each sample.The left side of the figure is the UPGMA tree of unweighted UniFrac distances between FH and AH (A) and FL and AL (B).Sequences were evenly pooled across individuals within a sample type prior to analysis.The right side of the figure is the principal coordinates analysis of an unweighted UniFrac distance matrix.AL:P.axillaris from low altitude; AH:P.axillaris from high altitude;FL:P.forsythii from low altitude; FH:P.forsythii from high altitude.

Table 1 Analysis of similarities of differences between groups.
3.3.Differential KEGG pathways among the gut microbiotaAfter classifying all KEGG Orthology (KO)into second-level functions,various KEGG pathways were significantly different in the intraspecific comparison of both species (Figure 5).InP.axillaris,carbohydrate metabolism,translation,cell motility,and lipid metabolism showed significantly higher levels in the microbiota of the high-altitude population.InP.forsythii,the genetic information process,metabolism of amino acids,and carbohydrate metabolism pathways were predicted at significantly higher levels in the microbiota of the high-altitude population.No significant difference in the abundance of KEGG pathways was detected in interspecific comparison at the high-altitude.In the interspecific comparison group at low-altitude,carbohydrate metabolism,unclassified metabolism,and protein families;signaling and cellular processes showed higher levels at highaltitude for both lizards and were predicted at significantly higher levels in the gut microbiota ofP.axillaris(Figure 5).

Figure 5 Different functions of gut microbiota of AL vs.AH and FL vs.FL.The microbial functions were predicted using PICRUSt at the second level of the KEGG pathway and were expressed as relative abundances.The differences between the levels of the predicted functions were tested using a two-sided Welch’s t-test,and P < 0.05 was considered as significant.AL:P.axillaris from low altitude; AH:P.axillaris from high altitude; FL:P.forsythii from low altitude; FH:P.forsythii from high altitude.
4.Discussion
Our results show neither heritable nor environmental factors are invariably dominant in determining the gut microbial communities ofP.axillarisandP.forsythii.Therefore,these results relate to the different local environmental stresses faced by the two lizards.
Environmental stress can also affect gut microbial communities,as the host and their microbes are considered as a co-evolved community (Shapira,2016).Hypoxia,a selective stress which can limit energy in high-altitude lizards (Yanget al.,2014a),causes intraspecific variation of the digestive tract structure (Hanet al.,2016) and affects gut microbial communities inPhrynocephalus vlangalii(Zhanget al.,2018).Thus,the environmental stress of limited energy may also select microbiota that can facilitate energy utilization in the host,leading to the convergence of gut communities of lizards at high-altitude (Yanget al.,2014a).
Our results indicate the gut microbiota ofP.axillarisandP.forsythiiat high elevation may promote energy utilization.For high-altitude lizards,increased fat utilization is an effective strategy for compensating the energy limitations caused by hypoxia (Tanget al.,2013).The abundance of Erysipelotrichia,which is negatively correlated with fat digestibility and protein metabolism (Spenceret al.,2011; Berminghamet al.,2017;Sunet al.,2018),was significantly higher in the low-altitude population ofP.axillaris.Furthermore,KEGG pathways related to carbohydrates and lipid metabolism showed significantly higher levels at high elevation,suggesting that there may be more energy produced (Burglinet al.,1987; Choiet al.,2015).Besides,amino acid metabolism by the microbiota in the animal intestine can ensure the host efficiently uses the amino acids available.Amino acids support the growth of gut microbiota and their host while regulating energy and protein homeostasis in the body (Daiet al.,2011; Yanget al.,2014b).These characteristics indicate that at high elevation,the gut microbes of the two lizards may facilitate greater energy production in their hosts.
At low-altitude,the different interspecific gut microbial communitiesmay result from different environmental stresses faced byP.axillarisandP.forsythii.High ambient temperature is an important form of environmental stress at low elevations(Sinervoet al.,2018),but may only affectP.forsythii.Our previous study indicated that ambient temperature was significantly negatively related to the genetic diversity ofP.forsythii(Qiet al.,2019) but positively related to the genetic diversity ofP.axillaris(Dinget al.,unpub.data).The relative high abundance of the pathogenic bacteria Rickettsiales,Anaplasmataceae,andWolbachia(Kocanet al.,2004) also indicated that high ambient temperatures reduce the survival status of low-altitude populations ofP.forsythii.Further,at low-altitude,P.axillarisandP.forsythiican be distinguished through their gut microbial community membership,which shows when environmental stresses differ for the two species,heritable factors may be dominant in determining the gut microbial communities.
Based on our results,heritable and environmental factors dominate the gut microbiota depending on the environmental stresses present.Additional experiments analyzing different environmental stresses are needed to clarify the role of environmental stress in determining the gut microbial communities.
Acknowledgements:The work was supported by the National Natural Science Foundation of China (No.31471988 and N0.31200287).We also thank You Li and Wen Zhong from Northwest Minzu University for their help with sampling.
 Asian Herpetological Research2020年4期
Asian Herpetological Research2020年4期
- Asian Herpetological Research的其它文章
- Characterization of Skin Symbiotic Bacteria of Sympatric Amphibians in Southeastern China
- Adaptive Evolution of the Ventral Scale Microornamentations among Three Snake Species
- Strong Limb Tactics of the Boulenger’s Lazy Toad,Scutiger boulengeri:Inferred from Limb Muscles
- Thermal Biology of Cold-climate Distributed Heilongjiang Grass Lizard,Takydromus amurensis
- The Relationship between Shell Morphology and Crevice Size Affecting Retreat Selection of the Keeled Box Turtle (Cuora mouhotii)
- Spring Voices in Korean Rice Fields:The Effect of Abiotic Variables and Syntopic Calls on the Calling Activity of the Treefrog Dryophytes suweonensis