
hTFtarget: A Comprehensive Database for Regulations of Human Transcription Factors and Their Targets
2020-11-06 01:03:32QiongZhangWeiLiuHongMeiZhangGuiYanXieYaRuMiaoMengxuanXiaAnYuanGuo
Qiong Zhang, Wei Liu, Hong-Mei Zhang, Gui-Yan Xie, Ya-Ru Miao,Mengxuan Xia, An-Yuan Guo,*
1 Department of Bioinformatics and Systems Biology, Key Laboratory of Molecular Biophysics of the Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan 430074, China
2 Clinical Center of Human Genomic Research, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China
KEYWORDS Transcription factor;ChIP-seq;Transcriptional regulation;Human;Database
Abstract Transcription factors (TFs) as key regulators play crucial roles in biological processes.The identification of TF–target regulatory relationships is a key step for revealing functions of TFs and their regulations on gene expression. The accumulated data of chromatin immunoprecipitation sequencing (ChIP-seq) provide great opportunities to discover the TF–target regulations across different conditions. In this study, we constructed adatabasenamed hTFtarget, which integrated hugehuman TF target resources (7190 ChIP-seq samples of 659 TFs and high-confidence binding sites of 699 TFs) and epigenetic modification information to predict accurate TF–target regulations. hTFtarget offers the following functions for users to explore TF–target regulations:(1)browse or search general targets of a query TF across datasets;(2)browse TF–target regulations for a query TF in a specific dataset or tissue;(3)search potential TFs for a given target gene or noncoding RNA; (4) investigate co-association between TFs in cell lines; (5) explore potential coregulations for given target genes or TFs; (6) predict candidate TF binding sites on given DNA sequences; (7) visualize ChIP-seq peaks for different TFs and conditions in a genome browser.hTFtarget provides a comprehensive, reliable and user-friendly resource for exploring human TF–target regulations,which will be very useful for a wide range of users in the TF and gene expression regulation community. hTFtarget is available at http://bioinfo.life.hust.edu.cn/hTFtarget.
Introduction
Transcriptional regulation is a fundamental and vital process for general and condition-specific gene expression [1]. Transcription factors(TFs)are the key regulators involved in transcriptional regulation [2]. Most TFs recognize and bind to specific DNA sequences named as transcription factor biding sites (TFBSs), leading to specific spatiotemporal expression patterns of target genes [3]. The disorder of TF–target regulation can disrupt normal biological processes,which may result in severe damage and diseases [4]. Thus, the identification of TF–target regulation is an important issue for understanding transcriptional regulation underlying complex biological processes [5]. Moreover, investigating spatiotemporal TF–target regulations can provide comprehensive insights into the regulatory mechanisms of gene expression in different cell status and diseases [6].
Chromatin immunoprecipitation sequencing (ChIP-seq)technology provides convenience to systematically investigate target genes of a TF at the genome-wide level[7].The accumulation of ChIP-seq data offers great opportunities to characterize the interactions between TFs and their targets in different conditions[8].Meanwhile,comprehensive utilization and visualization of TF–target data can offer systematic views for TF–target regulations.Although several resources,such as ReMap[9], CistromeDB [10], Factorbook [11], ChIPBase [12], and TRRUST [13], were conducted to display the relationships between mammalian TFs and their targets,most of them indirectly provided evidence for TF–target regulations.For example, ReMap and Factorbook present experimental designs of ChIP-seq datasets and peak information instead of putative TF–target regulatory relationships, while TRRUST deposits TF–target regulatory networks using a sentence-based text mining approach. Therefore, integrating large-scale omics datasets for TFs (including TFBSs, target prediction, mRNA profiling, and epigenetic status of chromatin) can provide a comprehensive resource of TF–target regulations and benefit researchers in transcriptional regulation studies [14].
In this study,we developed a database named hTFtarget by creating a comprehensive repertoire of TF–target relationships for humans,which offers an almost one-stop solution for studies involved in TF–target regulation. The hTFtarget has integrated thousands of ChIP-seq datasets and epigenetic modification information to predict reliable TF–target regulations, and also provides online tools to predict potential coassociation and co-regulation between TFs. In a word,hTFtarget can serve as a useful resource for researchers in the community of TF regulation and gene expression.
Implementation
hTFtarget was implemented with HTML, JavaScript (https://www.javascript.com/), Python (https://www.python.org/),Flask (http://flask.pocoo.org/), Bootstrap (https://getbootstrap.com/), AngularJS (a model-view-controller frame for Javascript web service,https://angularjs.org),and WashU Epi-Genome Browser[15].MongoDB(a cross-platform documentoriented database engine,https://www.mongodb.com/) was used to store metadata information. hTFtarget was hosted on the Ubuntu Linux system (version 16.04) with the Apache HTTP Server to provide a stable and open service.
Database content and methods
Data collection and quality control
Non-redundant ChIP-seq datasets of human TFs were curated from public databases, including NCBI Gene Expression Omnibus (GEO), NCBI Sequence Read Archive (SRA), and ENCODE[8].In GEO and SRA databases,ChIP-seq datasets were collected through the E-Utilities toolkit with filter criteria‘‘gds or sra, human orHomo sapiens,ChIP-seq or ChipSeq or ChIP sequencing,transcription factor or transcriptional factor or TF”. ChIP-seq datasets from ENCODE database were enrolled using parameters ‘‘assay_term_name = ChIP-seq,assembly=hg19/hg38,type=experiment,status=released,organism =Homo sapiens, target.investigated_as = transcrip tion factor”.All datasets were manually curated to discard the non-TF and abnormal datasets,such as artificial TFs(mutated or fused),transcriptional co-factors,general TFII family members, and unclear descriptions for experimental designs. After completing the aforementioned procedures, FastQC (v0.11.5)was used for data quality control (QC) to obtain clean reads.Bowtie (v1.2.1) was employed to align clean reads to human reference genome GRCh38. Datasets with <5 million reads after the QC procedure or an alignment ratio <50%were discarded. Finally, 7190 samples of 659 TFs from 569 conditions(399 cell lines, 129 tissues or cells, and 141 treatments) were kept for further analyses.
Peak detection and motif discovery
Bam files of technically replicated samples from the same dataset were merged together, and the input or IgG samples were used as controls according to the experiment design(IgG samples were used as controls only at the absence of input samples). The peak calling procedure followed the protocol of MACS2 (v2.1.0) pipeline with the following parameters (qvalue ≤0.01, fix-bimodal) [16]. Putative motifs of the TF in a dataset were identified using a similar method proposed by Wang and colleagues [17]. The detailed procedures are described as follows. (1) All of the peaks were ranked by the value of enrichment signal, and then the top 500 peaks (training set)were used for motif discovery using MEME-ChIP suite(v4.10.0) [18]. (2) The top five motifs (ranked by E-value,E-value ≤10-5and the ‘‘match sites” ≥100 in the top 500 peaks of step (1) were considered as confident ones, and the top 501–1000 peaks from the step 1 served as the testing set to measure the power of the top five motifs.The same number of random genomic regions of GRCh38 reference genome(non-peaks)with similar GC contents and length were selected as the control set. The FIMO (v4.10.0) [19] was used to scan both the testing and control sets, and then the numbers of recurrent motifs within the two sets were used to evaluate the significance of motifs for the TF (t-test with Bonferroni correctedPvalue <0.01).Motifs with significant reoccurrence only in the testing sets were considered as high-confidence ones and used for peak filtration. (3) Peaks with aPvalue ≤10-5and containing the aforementioned high-confidence motif(s)were considered as putative functional peaks and then used for further analyses.
Identification of TF–target regulation
Based on the putative functional peaks, we implemented the beta-model to identify candidate TF–target regulation [20],in which the beta-model score was used to measure the potential power of peaks for the TF–target regulation.
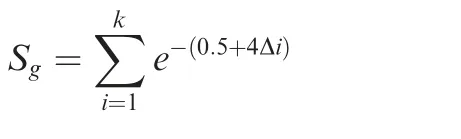
whereSgis the beta-model score represented as the sum of the weighted scores of peaks nearby the TSS of geneg, and the parameterkis the number of TFBSs within the 50 kb upstream from the TSS, while Δiis the distance between the summit of peakiand the TSS (which was normalized to 50 kb;e.g., the value 1 represents a 50 kb distance, while 0.04 indicates 2 kb).For an extreme case,only one peak was detected within 50 kb, and the summit of the peak was exactly at 2 kb upstream the TSS, the beta-model score was 0.517. Thus, the value 0.517 was used as the cutoff for regulatory capacity. If the putative functional peak with a beta-model score ≥0.517 locates within 50 kb upstream of the TSS, we consider the TF–target regulation is reliable. Further analyses and functional modules in hTFtarget were based on reliable TF–target regulation.
Moreover,we integrated TF–target regulations from multiple datasets of each TF and the chromatin status of the upstream region of a target gene, to survey whether the TF–target relationship is general or condition-specific. First, for each TF,we combined all of the targets of the TF from multiple datasets together, and the upstream 50 kb region from the TSS of each target was divided to subunits with a bin length of 200 bp. Then we examined whether any of the subunits was labeled with activated epigenetic modifications (data source curated from Roadmap project, http://www.roadmapepigenomics.org/). Finally, if the TF–target regulation is supported by one peak with a beta-model score ≥0.517 in more than 30%datasets of the TF,and any of the subunits of the region 50 kb upstream of the target gene is labeled with an activated epigenetic modification status within at least 30% of the Roadmap epigenomic samples,we consider the TF–target regulation is a general case; otherwise it is a condition-specific one.
Detection of TF co-association
The co-association of TFs, which was predicted according to the method proposed by Mark and colleagues [21], indicates the probability of TFs co-binding to common genomic regions.Briefly, to investigate the co-associations between a given TF(focus-factor)and other TFs(partner-factors)in a specific condition(a cell line or experiment),we first collected the overlapping regions of peaks between the focus-factor and each partner-factor to obtain a systematic co-binding map.We then implemented a machine learning method, which combined the RuleFit3 algorithm and mutual information of the overlapping regions between the focus-factor and partner-factors,to calculate the quantitative relationship as the relative importance score between the focus-factor and each partner-factor. A higher relative importance score of two given TFs indicates a stronger and more confident co-association relationship between them.
Co-regulation of TFs for the same target gene
Target gene co-regulated by more than two TFs was predicted based on the reliable TF–target regulations detected from curated ChIP-seq datasets in hTFtarget. Briefly, for a query gene, the region 50 kb upstream of the transcription start site(TSS) served as a core area to predict the candidate coregulation of TFs. TFs with high-confidence peaks within the region 50 kb upstream of TSS of target gene and the betamodel score of each peak ≥0.517 were considered as putative co-regulators to the query gene.
Sequence-based TFBS prediction
For the prediction of candidate TFBS(s)on given sequence(s),we integrated the motif matrices of humans from the hTFtarget database and other resources, including HOCOMOCO[22], JASPAR [23], and TRANSFAC v2018 databases [24].In total, we collected TFBSs for 864 human TFs, and the FIMO tool was employed for the TFBS discovery on given sequences.
Data resource and web-interface features
hTFtarget deposited 3.2 million records of TF–target regulations for 659 TFs in 569 experimental conditions and integrated 2737 high-confidence motifs of 699 TFs from other databases to predict 3.5 million records of candidate TF–target regulations. Moreover, 20,320 motifs were curated in the hTFtarget database from ChIP-seq data. Meanwhile, 408 TFs were found to possess co-association ability in 10 cell lines. Furthermore, hTFtarget provides a user-friendly web interface to facilitate search,browse,and download of comprehensive TF–target relationships in multiple experimental datasets,including putative TF–target regulations,ChIP-seq peaks,epigenetic modification status of TFBSs and targets, coregulation of TFs of the same targets, and co-association of TFs.All of the data sources and functional modules in hTFtarget are shown in Figure 1. We also compared hTFtarget with other resources related to TF–target regulation (Table 1). We found that hTFtarget may be the most comprehensive database providing various functions for the exploration of TF–target regulations in humans.
Usage

Figure 1 Overview of data resources and functional modules of hTFtarget
First, hTFtarget provides a ‘‘Quick Search” function on the top right of each page for users to conveniently survey the TF–target regulations. Various types of inputs are acceptable,such as the gene name,the Ensembl gene ID,gene symbol,and alias. Users can obtain basic information for the query TF or gene, related TF–target regulations, epigenetic modification status in the promoter region of target gene, and detailed evidence for the TF–target regulation in different conditions.For example, when inputting geneASCL2on the ‘‘Quick Search”function(Figure 2),all records ofASCL2as a TF gene or a target gene are shown in Figure 2A.
On one hand,when the query geneASCL2is a TF gene,the‘‘Details”icon below the term‘‘Targets”(red box in Figure 2A)links to a page displaying its targets with three levels of evidence (Figure 2B). Additionally, users can filter target genes by support levels (‘‘Epigenetic Evidence”, ‘‘ChIP-seq Evidence”,and‘‘Motifs Evidence”)and browse the peak information through the ‘‘view” icon (Figure 2C). On the other hand,when the query geneASCL2is a target gene, the ‘‘Details”label underlying the term‘‘TFs”(blue box in Figure 2A)shows all records about which TFs could regulate the target geneASCL2(Figure 2D). Users can browse the peak information for the TF–target regulation by clicking the ‘‘Details” icon(Figure 2E).
Other than the quick search function, hTFtarget offers the following six modules for users to conveniently explore TF–target regulations.
TF module
The TF module provides details of TF–target relationships and ChIP-seq experimental designs (Figure 3A and B). Users can explore the general(Figure 3A)or condition-specific TF–target regulations (Figure 3B), as well as browse the TF annotation(the bottom of Figure 3A) and the experimental design of the interested dataset (the bottom of Figure 3B). The ‘‘details”icons in Figure 3A enable users to browse the detailed information of TF–target regulations(Figure 3C),including the epigenetic status of the upstream region for the target gene and the visualized peak information in the genome browser (Figure 3D). Additionally, beyond surveying TF–target regulation in hTFtarget online, users can download the records as well.

Target module
The target module helps users to investigate the TF(s) that regulate(s) the query gene. The function and the interface of this module are similar to what are shown in Figure 2D and 2E.
Peak module
The peak module provides comprehensive information of peaks in a user-defined manner (Figure 4A). Users can browse the peaks of different TFs in the selected condition(e.g.,the same cell line or tissue)or investigate TF–target regulations for the same TF in different conditions(e.g.,the distinct spatiotemporal TF–target regulatory relationships in different tissues).
Co-regulation module
The co-regulation module helps users to search the common targets that can be regulated by the given TFs,or search common TFs that can regulate the given genes (Figure 4B).
Co-association module
The co-association module predicts and visualizes potential co-association TFs in the collected cell lines. The coassociation of two TFs indicates the probability of their combinatorial occupancies on the same genomic regions. The human TFs have always shown distinct co-association relationships in combinatorial and context-specific patterns for gene regulation [21,25]. The relative importance score indicates the probability of co-association between two TFs,and the higher score means stronger co-association in the corresponding condition.
Prediction module
The prediction module employs motif matrices to predict potential TFBSs on the user-provided sequence(s) (Figure 4C).The motif matrices were curated from TRANSFAC,JASPAR, HOCOMOCO, and hTFtarget databases.
Discussion
Understanding TF–target regulatory relationships is a critical issue when investigating complex molecular mechanisms underlying biological processes and diseases [26,27]. Up to date, several resources have been dedicated to identify TFs,such as PlantTFDB and AnimalTFDB [28,29], and largescale of ChIP-seq data have been available for the detection of TF–target regulatory relationships as well. Integrative resources would offer significant insights into the dynamic TF–targets regulatory mechanisms[30].In this study,we presented the hTFtarget, a comprehensive resource focusing on the regulation between human TFs and their targets by integrating ChIP-seq data and TFBS prediction.

Figure 2 Snapshots of the quick search function in hTFtarget
Although several resources could indirectly investigate potential regulatory interaction between TF(s) and target(s)based on ChIP-seq peak signals,a database concisely indicating target genes for specific TFs and revealing comprehensive regulations of TF–targets is still lacking. For example,although a large number of datasets and TFs has been collected in CistromeDB,it does not distinguish TFs,transcriptional cofactors,RNA polymerases,TFII family members,or their compounds.Additionally,although CistromeDB provides a search function for TF–target regulations of a single query gene and browse peak information of a given TF, it ignores the coregulation information for TF–targets, and does not refer to the TF–target regulations at the genome-wide level. The ReMap database just provides peak information for TFs in ChIP-seq datasets. The HOCOMOCO [22], JASPAR [23],TRANSFAC v2018 [24], and TFBSbank [31] databases focus on the collection and discovery of the motif profiles for TFBSs,while TRRUST elucidates TF–target interactions using text mining[13].hTFtarget is the only currently available database integrating various resources to provide an almost one-stop solution in investigating TF–target regulations in humans.
Further development of hTFtarget will be focused on spatiotemporal classification of TF–target regulations in different gene families and diseases,such as integrating with GSCALite cancer analysis[32].Additionally,we will continue to upgrade hTFtarget with new datasets and more epigenetic modifications,keep improving algorithms for better accuracy,and offer more functions and tools. We will maintain and regularly update hTFtarget as we do for our AnimalTFDB database,which have been maintained for more than eight years. We believe that hTFtarget will be a valuable resource for the research community.
Conclusion
The identification of TF–target regulations is an important issue for revealing the molecular mechanisms underlying complex biological processes. Huge amount of ChIP-seq resources and epigenetic modification information have been integrated into hTFtarget to identify putative TF–target regulations in humans. hTFtarget provides a comprehensive, reliable, and user-friendly resource, and an almost one-stop solution to explore TF–target regulations in humans.

Figure 3 Views for the TF and target related modules in hTFtarget
Data availability
hTFtarget is freely accessible at http://bioinfo.life.hust.edu.cn/hTFtarget/.
CRediT author statement
Qiong Zhang:Methodology, Software, Data curation,Resources, Formal analysis, Visualization, Writing - original draft, Writing - review & editing, Funding acquisition.Wei Liu:Methodology, Software, Data curation, Formal analysis,Visualization.Ya-Ru Miao:Formal analysis.Mengxuan Xia:Formal analysis.Hong-Mei Zhang:Methodology, Software,Data curation,Formal analysis.Gui-Yan Xie:Formal analysis.Ya-Ru Miao:Formal analysis.Mengxuan Xia:Formal analysis.An-Yuan Guo:Conceptualization, Project administration,Funding acquisition, Writing - review & editing. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.

Figure 4 Other important modules in hTFtarget
Acknowledgments
We would like to thank colleagues in groups of Ensembl,GEO, SRA, ENCODE, TRANSFAC, JASPAR, HOCOMOCO, Roadmap Epigenomics, and AnimalTFDB. We also acknowledge the funding from the National Natural Science Foundation of China (Grant Nos. 31822030, 31801113, and 31771458), National Key R&D Program of China (Grant No. 2017YFA0700403), and China Postdoctoral Science Foundation (Grant No. 2018M632830).
ORCID
0000-0001-5001-8434 (Zhang, Q)
0000-0001-9881-785X (Liu, W)
0000-0001-9835-9684 (Zhang, HM)
0000-0002-2924-5306 (Xie, GY)
0000-0002-1982-3141 (Miao, YR)
0000-0002-8270-4444 (Xia, M)
0000-0002-5099-7465 (Guo, AY)
 Genomics,Proteomics & Bioinformatics2020年2期
Genomics,Proteomics & Bioinformatics2020年2期
- Genomics,Proteomics & Bioinformatics的其它文章
- SuccSite: Incorporating Amino Acid Composition and Informative k-spaced Amino Acid Pairs to Identify Protein Succinylation Sites
- HybridSucc: A Hybrid-learning Architecture for General and Species-specific Succinylation Site Prediction
- BGVD: An Integrated Database for Bovine Sequencing Variations and Selective Signatures
- SR4R: An Integrative SNP Resource for Genomic Breeding and Population Research in Rice
- IC4R-2.0: Rice Genome Reannotation Using Massive RNA-seq Data
- iGMDR: Integrated Pharmacogenetic Resource Guide to Cancer Therapy and Research