
Cu2O 微米晶的调控合成及影响其形貌的因素
2020-09-28 09:19顾修全倪中海应鹏展
实验技术与管理 2020年5期
张 伦,顾修全,倪中海,应鹏展
(1.中国矿业大学 材料与物理学院,江苏 徐州 221116;2.中国矿业大学 化工学院,江苏 徐州 221116;3.中国矿业大学 徐海学院,江苏 徐州 221116)
氧化亚铜(Cu2O)是一种p 型直接带隙半导体,禁带宽度约为1.9~2.2 eV[1],因其在许多领域具有潜在应用价值而备受研究者的青睐。例如,Cu2O 可用作太阳能转换材料[1]、润滑剂[2]、气敏材料[3]、磁存储材料[4]、锂电极负极材料[5]和光催化剂[6]等。研究者们主要从制备方法、形貌调控和性能研究等方面对Cu2O展开了广泛的研究。Cu2O 的制备方法多种多样,有(水热)溶剂热法[7]、电沉积法[8]、化学沉淀法[9]、原位沉淀法[10]、超声制备法[11]、种子生长法[12]和物理气相沉积法[13]等。由于不同形貌的Cu2O 微纳米晶体具有不同的性能,如Cu2O 颗粒越小,比表面积越大,其光催化活性、气敏性以及吸附性能等越强。因此,调控合成不同尺寸和结构的Cu2O 微纳米晶是材料科学者多年来研究的热点内容。总的来说,Cu2O 晶体的形貌调控包含3 类常见的方法:1)改变溶液的pH 值和浓度以及反应时间和反应温度[14-18];2)使用不同的表面活性剂或分散剂作为软模板,如聚乙烯吡咯烷酮(PVP)、十二烷基硫酸钠(SDS)和十六烷基三甲基溴化铵(CTAB)[19-22]等;3)采用不同的添加剂和还原剂,如水合肼(N2H4·H2O)和硼氢化钠(NaBH4)等[1]。
本文采用化学沉淀法,以硫酸铜(CuSO4)作为铜源,分别用水合肼(N2H4·H2O)和抗坏血栓(C6H8O6)作为还原剂,采用PVP 为表面活性剂,调控合成不同结构和尺寸的Cu2O 微米晶。主要探讨了表面活性剂PVP 的用量以及反应温度对Cu2O 微米晶形貌的影响。并且将PVP 的用量对Cu2O 微米晶形貌的影响与纳米晶进行了对比,尝试从微米颗粒和纳米颗粒受力的角度,分析了PVP 用量对Cu2O 微米晶和纳米晶不同的作用效果,为基础研究提供一些基本实验数据。
1 实验方法
1.1 样品的制备
1.1.1 样品S1 和S2 的制备
所有样品均是分析纯,使用时没有进一步做纯化处理。参考文献[23],适当改进合成方法,进行样品合成。制备样品的具体过程是:配制100 mL 浓度为0.01 mol/L 的CuSO4水溶液,加入单口圆底烧瓶内,将其水浴加热至20 ℃(室温约5 ℃);接着向其中加入0.180 g PVP( K30),搅拌至溶解;然后缓慢逐滴滴加配制好的25 mL NaOH 水溶液(1.5 mol/L),很快有蓝色沉淀生成;继续搅拌1~2 min 后,缓慢滴加大约2 mL 的水合肼的水溶液(1∶15),颜色逐渐由蓝色变为血红色,搅拌30 min 后高速离心,用去离子水和无水乙醇交叉洗涤数次,即得到Cu2O 颗粒。最后将合成的Cu2O 颗粒在60 ℃下真空干燥4 h,此样品编号记为S1。将PVP 的用量改为0.338 g,其他合成条件和过程与S1 相同,这样合成的样品记为S2。
1.1.2 样品S3 和S4 的制备
其他合成条件和过程与S1 相同,仅将2 mL 的水合肼的水溶液用25 mL 0.1 mol/L 的抗坏血酸水溶液替换,溶液则很快由蓝色变为橙红色,合成的样品记为S3。保持其他条件和过程与S3,将PVP 的用量改为0.338 g,合成的样品记为S4。
1.1.3 样品S5-S7 的制备
其他合成条件和过程与S1 相同,仅将水浴温度提高至60 ℃,合成的样品记为S5。其他合成条件与过程与S3 相同,仅将水浴温度提高至50 ℃,合成的样品记为S6。其它合成条件和过程与S6 相同,仅将抗坏血酸的用量加倍,合成的样品记为S7。
合成的7 个样品的实验条件和形貌如表1 所示。其中,合成每个样品时,CuSO4溶液和NaOH 溶液的用量和浓度均保持不变,且各样品的反应时间均为30 min。
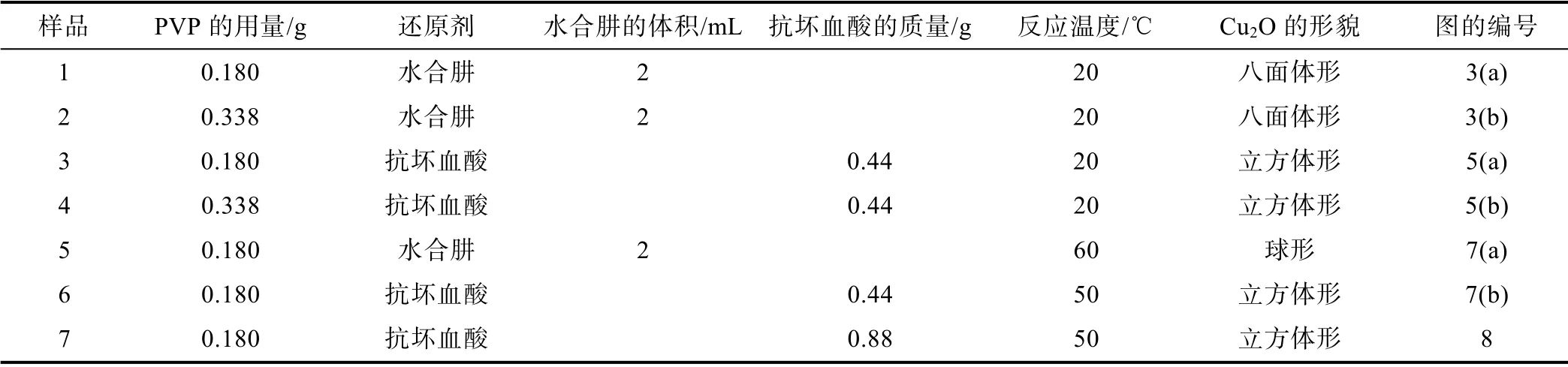
表1 样品形貌和制备条件小结
1.2 样品测试
用德国布鲁克D8 ADVANCE 型X-射线衍射仪(XRD)测试样品的纯度和晶体结构等。本实验所用的阳极金属靶为Cu 靶,X-射线管的电压和电流分别是40 kV 和30 mA,X-射线波长 λ =1.5418Å。用日本日立S-4800 型场发射扫描电子显微镜(FESEM)测试样品的形貌,加速电压为5 kV。用美国飞雅(FEI)Tecnai G2 F20 型场发射透射电子显微镜(TEM)高分辨透射电镜分析样品的形貌和结构,加速电压为200 kV。用美国尼高力Nicolet iS5 型傅立叶变换红外光谱仪(FT-IR)测试样品的红外光谱,采用掺入一定量的Cu2O 粉末的KBr 压片进行测试,其中波数范围为350~800 0 cm-1。
2 实验结果与分析
2.1 X-射线线衍射分析
在合成的7 个样品中,仅对Cu2O 八面体(S1)和Cu2O 立方体(S3)2 个样品进行了XRD 表征,因为其他几个样品或是形貌较S1(S3)没有明显改变或是形态呈无定型。图1 给出了不同形貌的Cu2O 晶体的XRD 衍射谱图,可以看出每个衍射峰的峰位和相对强度大小。对照国际晶体衍射卡JCPDS No.05-0667号,得知制备的Cu2O 晶体属于立方晶系。5 个最强衍射峰分别对应晶面指数为(110)、(111)、(200)、(220)和(311)的晶面族。由图1 可见,每个样品已经结晶,但是,衍射图谱中的(110)衍射峰左侧还有一个衍射峰。查考资料得知,此峰既不是CuO 晶体的某个晶面族的衍射峰[24],也不属于Cu 单质的某个晶面族的衍射峰[18],分析认为此峰很可能是未清除干净的极少量杂质的衍射峰。而且,后面即将讨论的FT-IR 光谱测试表明合成样品的确为Cu2O 晶体。
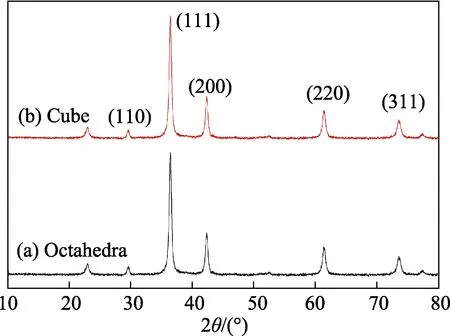
图1 Cu2O 八面体(a)和Cu2O 立方体(b)的XRD 衍射谱图
2.2 红外光谱分析
同样,在合成的7 个样品中,仅对Cu2O 八面体(S1)和Cu2O 立方体(S3)2 个样品进行了FTIR 测试,以便与XRD 测试结果比较。用红外光谱来检测Cu2O 八面体和Cu2O 立方体的纯度,测试结果如图2所示。测试样品时,扣除了空气背底,Cu2O 八面体的红外光谱谱图中只含有位于632 cm-1的Cu-O 伸缩振动吸收峰。而Cu2O 立方体在3 447 cm-1处有很微弱的吸收,可能是纳米粒子对空气中的水(O-H)具有较强的吸收作用所致[25-26],虽然测试样品时用红外灯进行了烘干处理,但未能全部去除水分。FTIR 测试表明2 个样品均不含杂质,这与前面的XRD 分析基本一致(XRD 谱图中还有一个非常微弱的杂质衍射峰,红外光谱没有检测出来)。

图2 Cu2O 八面体(a)和Cu2O 立方体(b)的红外谱图
2.3 表面积活性剂PVP 用量对样品形貌的影响
采用水合肼作为还原剂,当PVP 的用量为0.180 g时,合成得到样品S1。S1 的SEM 图像如图3(a)所示,为形貌规整的Cu2O 八面体,棱角分明,颗粒直边边长约为700~900 nm,且尺寸分布比较均一。增加PVP的用量为0.338 g,合成的样品S2 仍然为八面体形(见图3(b)),颗粒尺寸和均匀性没有明显变化,但可以看出,样品S2 的表面比样品S1 的表面略微光滑。分析认为,PVP 的用量增加可能会阻止液相中的Cu2O 小颗粒附着在大颗粒上,从而使得样品S2 的每个表面变得更加光滑,棱角更加明锐。图4(a)给出了样品S1 的TEM图像,样品尺寸与横断面与SEM 的测试结果一致。

图3 采用水合肼为还原剂,在不同PVP 的用量下合成的Cu2O 八面体的SEM 图像

图4 使用不同还原剂,合成得到的Cu2O微米晶的TEM 图像
采用抗坏血酸为还原剂,当PVP 的用量为0.180 g时,合成得到样品S3。S3 的SEM 图像如图5(a)所示,合成的Cu2O 微米晶有的棱角分明,有的棱角稍微圆润一些,但都是Cu2O 立方体,棱边边长约为300~600 nm,尺寸的均一性比Cu2O 八面体的略微差一些。随着PVP 用量增加至0.338 g,合成的样品S4 仍然为立方体形(见图5(b))。与样品S3 相比,样品S4 中的Cu2O 大立方块的数目略有减小,而小立方块的数目有所增加。分析认为,PVP 的增加可能会阻止Cu2O立方体颗粒继续长大,所以小颗粒明显增加。这一点,有别与上述的Cu2O 八面体,可能因Cu2O 颗粒越小,其形貌受到PVP 用量的影响越大。后面将进一步分析。图4(b)给出了样品S3 的TEM 图像,样品尺寸与横断面与SEM 的测试结果一致。
由上述讨论可知,PVP 的用量在0.180~0.338 g范围内变化时,无论是Cu2O 八面体,还是Cu2O 立方体,其形貌和分散性等都没有很明显的变化,这一结果有别于PVP 的用量对Cu2O 纳米晶的影响。本团队此前合成了平均粒径为25 nm 的Cu2O 纳米晶,讨论了PVP 用量对其形貌的影响,如图6 所示[27]。不添加PVP 时,Cu2O 呈无定形生长,并且颗粒团聚很严重,如图6(a)所示。当PVP 用量增加至0.165 g 时,合成得到分散性较好的类球形Cu2O 纳米晶,如图6(b)所示,分析认为由于大量PVP 分子包裹在Cu2O 纳米颗粒的表面,阻止了小颗粒的继续团聚和长大。而当PVP用量继续增加到0.265 g 时(见图6(c)),Cu2O 颗粒形貌和大小虽然没有明显变化,但颗粒团聚再次发生,形成许多“胶团”,这可能是由于PVP 的浓度超过临界胶团浓度所致。总之,当PVP 的用量改变相近时,所引起的Cu2O 微米晶的形貌改变要远小于纳米晶。就其原因,下面将从Cu2O 微米晶和纳米晶受力的角度,进行粗略浅显地探讨。

图5 采用抗坏血酸为还原剂,在不同PVP 的用量下合成的Cu2O 立方体的SEM 图像

图6 Cu2O 纳米晶在高倍下的SEM 图像[27]
晶体生长取向连接(OA)理论的创始人Banfield等[28]提出晶体生长与微观粒子受力有关。他们首先跟踪拍摄了2 个零维(0D)纳米颗粒的连接过程,然后计算出纳米颗粒的加速度a,根据牛顿第二定律F=ma(F,m 分别表示纳米颗粒所受的合力及质量),计算其所受合力大小,并分析认为纳米颗粒所受合外力是颗粒间引力和斯托克斯阻力2 个力的合力。然而,本文涉及的制备体系以及相关的实验设备,还不具备条件来定量计算微米颗粒的受力大小,甚至还不能分析清楚一个微米颗粒受到哪些力的作用。但可以看出,即在足够小的力的作用下,纳米颗粒的形貌和分散性等很可能发生变化;而微米颗粒只有受到最够大的力的作用,形貌才可能发生变化。当PVP 用量改变相近时,Cu2O 纳米球的形态和分散性有很大改变;而Cu2O八面体(立方体)形态没有明显改变。至于这里讨论的PVP 的用量需要增加多少,Cu2O 微米晶的形貌才可能发生变化,还需要系列实验来进一步探讨。
2.4 反应温度对样品形貌的影响
为了研究反应温度对Cu2O 微米晶最终形貌的影响,将反应体系的温度提高至60 ℃和50 ℃,分别得到样品S5 和S6,其SEM 图像如图7 所示。比较图3(a)和图 7(a)可见,随着合成温度从 20 ℃提高到60 ℃,Cu2O 八面体(S1)由原来的八面体形演变为类球形(S5),且大部分颗粒尺寸没用明显的变化,只是有极少量的粒径为200~400 nm 的小球出现。究其原因,可能与升温过程中蓝色的Cu(OH)2受热分解为黑色的CuO 有关。实验中发现,20 ℃下合成样品时,没有经历CuO 颗粒生成的这一环节;而60 ℃下合成样品的过程中,经历了蓝色颗粒变为黑色颗粒的过程,这一变化显然是Cu(OH)2颗粒受热分解为CuO颗粒。因此,样品S5 有待更多的检测和分析。

图7 在60 ℃(球)和50 ℃(立方体)下合成样品的SEM 图像
考虑到抗坏血酸的性能在高温下可能受到影响,合成样品时未将水浴温度调至 60 ℃,而只是调至50 ℃,合成的Cu2O 立方体(S6)如图7(b)所示。比较图3(b)和图7(b)可见,随着合成温度从20 ℃提高到50 ℃,Cu2O 立方体(S6)的每个表面附着了许多小颗粒,这一结果有别于表面光滑的Cu2O 立方体(S3),但立方体颗粒大小并没有明显的改变,其深层原因有待进一步的探讨。为了排除抗坏血酸的性能在高温下变差的可能性,造成了Cu2O 晶粒的最终形貌改变。将抗坏血酸的用量由原来的0.44 g(S6)提高一倍为0.88 g,保持实验其他件都不变,继续进行了实验研究,这样合成的样品为S7,其SEM 图像如图8 所示。与样品S6 的SEM 图片(图7(b))相比,图8 所示颗粒形貌也没有明显改变,只是颗粒尺寸略显均匀。由此可见,在20 ℃和50 ℃下合成的样品(S3 和S6)在形貌上的差异可能不是抗坏血酸的性能在高温下变差所致,而是反应温度本身变化引起。

图8 在50 ℃下,使用0.88 g 抗坏血酸合成样品(S7)的SEM 图像
3 结语
本文合成了Cu2O 八面体和立方体等7 个微米晶样品,探讨了表面活性剂PVP 的用量和反应温度等因素对Cu2O 微米晶最终形貌的影响,主要得到如下结论:当PVP 的用量由0.180g 增加至0.338 g 时,Cu2O八面体和Cu2O 立方体的形貌变化不大,PVP 的用量对Cu2O 微米晶形貌的影响远不及对纳米晶的影响大。采用水合肼作为还原剂时,随着水浴温度从20 ℃增加至60 ℃,Cu2O 微米晶的形貌从八面体演变为球状。而采用抗坏血酸作为还原剂时,当水浴温度从20 ℃增加至50 ℃,Cu2O 微米晶的形貌没有明显变化,仍为立方体形状,只是在立方体上附着了很多小颗粒。
猜你喜欢
氯碱工业(2022年6期)2022-11-21
教学考试(高考化学)(2022年4期)2022-08-30
食品安全导刊(2022年2期)2022-03-18
陶瓷学报(2021年3期)2021-07-22
化工环保(2021年2期)2021-04-25
上海师范大学学报·自然科学版(2020年2期)2020-12-28
云南化工(2020年5期)2020-06-12
新世纪智能(数学备考)(2019年9期)2019-10-16
中国蔬菜(2016年1期)2016-05-26
中国粮油学报(2016年1期)2016-02-06
