
气相色谱–质谱法快速筛查农药乳油制剂中非法添加物
2020-09-26 08:16:08王婷李建兵赵维一李磊王光英丁白瑜王晓滨
化学分析计量 2020年5期
王婷,李建兵,赵维一,李磊,王光英,丁白瑜,王晓滨
(1.青岛市产品质量监督检验研究院,国家海洋精细化工及其生物制品质量监督检验中心,山东青岛 266101;2.青岛市城阳区市场监督管理局,山东青岛 266109; 3.山东省农药科学研究院,济南 250100)
根据农药登记情况,我国目前使用较多的是化学农药。农药原药不能直接使用,要加工成特定的剂型应用于农业生产。在农药产品中乳油制剂约占25%,使用率很高[1]。从2005 年开始,我国成为世界第一农药生产国和出口国[2],农药产品质量持续受到人们的关注。2018 年农业部关于农药产品抽查结果显示,农药产品质量不合格样品551 个,其中非法添加其它成分的样品222 个,占农药不合格产品的40%,对农药产品质量安全产生重要影响。因此需要建立一种快速、准确、灵敏筛查农药乳油制剂中非法添加农药的方法。
目前,农药成分检测方法有气相色谱(GC)法[3]、液相色谱(LC)法[4]、气相色谱–质谱联用(GC–MS)法[5~6]、液相色谱–质谱联用(LC–MS/MS)法[7]、液相色谱–飞行质谱(LC–Q–TOF)法[8]。GC 和LC 法主要依据保留时间定性,但是具有相同保留时间的物质不一定是同一化合物,尤其LC 法很难保持仪器条件完全相同,可能导致保留时间漂移,仅靠保留时间定性化合物容易出现误判[9]。质谱通过分子离子峰和碎片离子峰得知化合物有关分子结构的信息,对化合物的定性更加准确。LC– MS/MS 法适用于极性和不稳定性农药的测定。LC–Q–TOF 法设备昂贵,农药制剂中农药浓度高,易污染仪器,影响仪器的残留检测,检测成本高。而GC–MS 法具有GC 法的高分离度和MS 法的定性优势,可以准确定性定量分析多种农药成分[10]。
笔者建立了农药乳油制剂产品中54 种常见杀虫剂的GC–MS 快速筛查方法,并应用于实际农药产品的验证。
1 实验部分
1.1 主要仪器与试剂
气相色谱–质谱联用仪:Agilent 5975B +6890N型,美国安捷伦科技有限公司;
电子天平:(1)ML204T/02 型,感量为0.1 mg,(2)MS205DU 型,感量为0.01 mg,瑞士梅特勒–托利多国际贸易(上海)有限公司;
甲苯、丙酮:均为色谱纯,美国Anaqua Chemicals Supply 公司;
敌敌畏等53 种农药标准样品:德国Dr. Ehrenstorfer 公司,各标准样品的纯度见表1;

表1 农药标准样品纯度
腈菌唑、嘧菌酯标准样品:纯度均为100%,美国Accustandard 公司。
1.2 仪器工作条件
1.2.1 气相色谱
色谱柱:DB–17MS 毛细管柱(30 m×0.25 mm,0.25 μm,美国安捷伦科技有限公司);进样口温度:290℃;载气:He,流量为1.0 mL/min;进样体积:1 μL;进样方式:分流进样,分流比为10∶1;程序升温条件:40℃保持1 min,以30℃/min 升至140℃,保持1 min,再以5℃/min 升至250℃,保持5 min,最后以10℃/min 升至280℃,保持5 min。
1.2.2 质谱
离子源温度:230℃;四级杆温度:150℃;接口温度:280℃;电离方式:EI 源;扫描方式:全扫描(SCAN)和选择离子监测(SIM)。
1.3 标准溶液的配制
1.3.1 农药单成分标准储备液
准确称取10 mg(精确至0.01 mg)各农药标准样品于10 mL 容量瓶中,用甲苯配制成1.0 mg/mL的标准储备液。
1.3.2 混合农药标准溶液
准确移取不同体积的各农药单成分标准储备液于10 mL 容量瓶中,用丙酮稀释成各组分质量浓度均为10 mg/L 的混合农药标准溶液。
1.4 样品前处理
称取0.1 g 样品于10 mL 容量瓶中,加入5 mL丙酮,溶解,再用丙酮定容至标线,混匀,过滤,待测。
2 结果与讨论
2.1 农药品种的选择
根据农业部发布的近几年关于农药监督抽查的结果,非法添加的农药主要包括禁限用农药如水胺硫磷、氟虫腈、克百威、硫丹等;高活性过专利保护期农药如螺螨酯、氯虫苯甲酰胺、虫螨腈等;速效性或活性较好的农药如菊酯类、阿维菌素、甲氨基苯甲酸盐,毒死蜱等[11]。
甲氨基阿维菌素苯甲酸盐、氯虫苯甲酰胺等农药一般采用高效液相色谱法分析,不适合用气相色谱分离,这类农药暂未列入考察范围。在前期预实验过程中,发现螺螨酯和百菌清在空白乳油样品基质中的回收率很低。根据中国农药信息网,螺螨酯和百菌清没有乳油制剂产品的登记记录,这可能是由于螺螨酯和百菌清在乳油制剂中溶解性不好,不适合做乳油制剂,也未列入考察范围。结合近几年农业部、前工商系统、质检系统各级监督抽查结果和检测机构检验结果,选取54 种常见的农药作为研究对象。
2.2 提取溶剂的选择
乳油是原药溶解在有机溶剂中,如甲醇、乙腈、丙酮、苯系物等,并加入一定量的专用乳化剂而制成的[12],丙酮与常见乳油溶剂具有良好的混溶性,因此选取丙酮作为溶剂提取样品中的农药成分。
2.3 基质效应
基质效应反映了样品中其它成分对待测物测定的影响,是农药分析时需要考虑的一个重要问题,通常用基质效应因子(MF)来表示[13–14]。GC–MS 法的基质效应一般以增强为主,这可能是由于基质的保护作用,与纯溶剂相比,空白基质溶液可以使待测物更加完全地转移至色谱柱中,从而响应增大[15]。
分别用空白基质溶液和纯溶剂配制0.1~4.0 mg/L 的混合标准工作溶液,分别测定并建立标准工作曲线,通过比较两条标准工作曲线的斜率分析基质效应(MF)的强弱:
MF=[(基质曲线斜率/溶剂曲线斜率) –1]×100%
结果表明,11%的农药出现了强基质效应(MF>50%),因此需要用不含待测物农药的空白基质溶液配制标准溶液,在一定程度上可减弱基质效应对目标化合物测定的影响。
2.4 GC–MS 分析条件
采用气相色谱法检测农药主要用非极性、弱极性到中等极性的色谱柱,其中HP–1 型、HP–5 型非极性或弱极性色谱柱主要用于分离有机氯、菊酯类农药,在分离有机磷农药时会出现拖尾;有机磷类极性农药更多使用中等极性的DB–17 型、DB–1701型色谱柱。本研究项目涉及的农药种类多,包含有机磷、有机氯、菊酯类等不同类型农药,农药极性和沸点差异较大,因此选用DB–17MS 柱分离待测物,在分离过程中个别农药没有实现基线分离,如丙环唑和炔螨特,但是由于它们之间没有共同的特征离子,不影响检测。
色谱有冷柱头进样、填充柱进样、分流/不分流进样方式,实验室只有分流/不分流进样口,故选择分流/不分流进样方式。不分流进样主要用于痕量农药残留的测定,本项目的检测对象是农药制剂中含量高于0.1%的非法添加农药成分;另外不分流进样时,高浓度待测成分可能污染进样口、质谱离子源甚至四极杆检测器。因此选择分流进样,一方面可以避免污染色谱和质谱仪,另一方面分流进样方法的定量限远低于0.1%(1 000 mg/kg),此外分流进样色谱峰形也更好。
本研究项目涉及的农药种类比较多,沸点范围大,譬如苯醚甲环唑(480℃)、烯酰吗啉(372℃)、嘧菌酯(581℃)、唑虫酰胺(540℃)等农药的沸点很高,为提高待测农药在进样口的气化效率,选择较高的进样口温度(290℃)。若采用温度较低的恒温程序,则沸点较高的农药不易出峰,甚至残留在色谱柱中,出现假阴性或者不能检出;若采用温度较高的恒温程序,则沸点较低的农药分离度较低,会出现色谱峰重叠,分析灵敏度降低。为提高待测农药的分离度,选择程序升温,以使尽可能多的农药组分基线分离[6]。
2.5 线性关系和检出限。
用空白基质溶液配制0.1~4.0 mg/L 的系列标准工作溶液,按1.2 仪器工作条件进行分析,混合农药总离子流色谱图如图1 所示。由图1 可知,各农药色谱峰可有效分离。
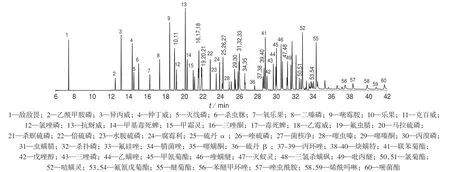
图1 54 种农药总离子流色谱图
以各农药的质量浓度为自变量、对应色谱峰的峰面积为纵坐标进行线性回归,计算线性方程和相关系数。
以定量离子的3 倍和10 倍信噪比分别计算各农药测定方法的检出限(LOD)和定量限(LOQ)。
54 种农药的线性方程、相关系数、检出限和定量限列于表2。

表2 54 种农药的线性方程、相关系数、检出限与定量限
由表2 可知,54 种农药的线性相关系数(r2)均大于0.990,表明在0.1~4.0 mg/L 质量浓度范围内,标准工作曲线线性良好。54 种农药的检出限为0.05~4.99 mg/kg,定量限为0.15~16.63 mg/kg。
2.6 加标回收试验
对乳油空白样品进行加标回收试验,选择10.0 mg/kg 和20.0 mg/kg 两个加标水平,每个水平重复测定6 次,测定结果列于表3。由表3 可知,54 种农药的加标回收率为83.7%~129.3%,测定结果的相对标准偏差为0.8%~16.5%,本法精密度、准确度满足分析要求。

表3 54 种农药的加标回收试验结果 %
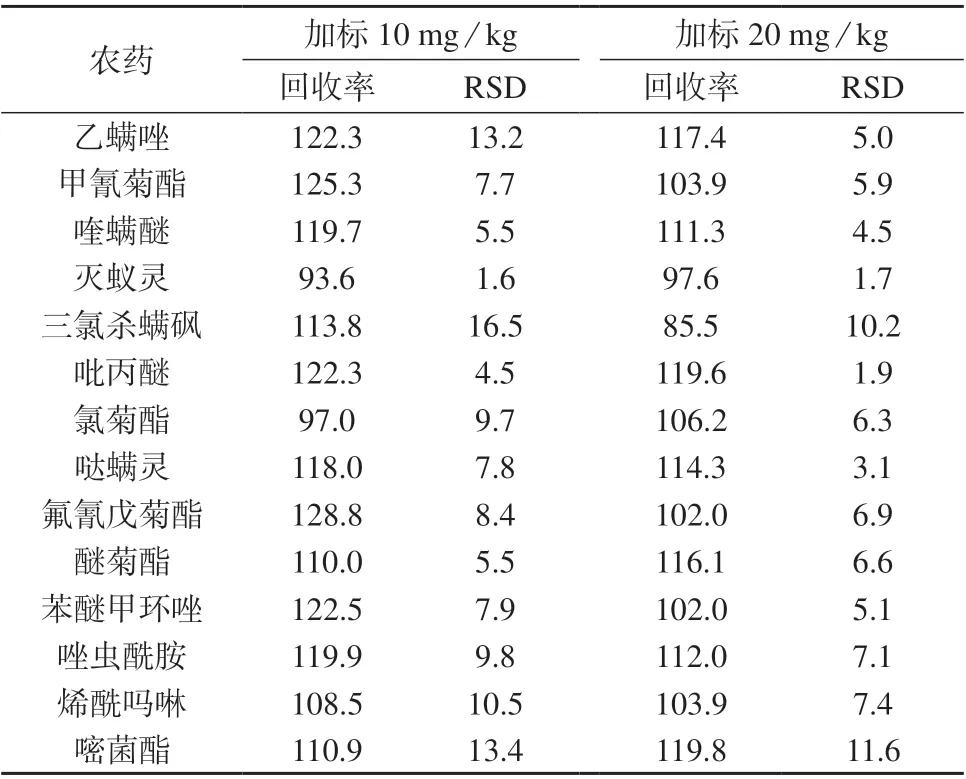
续表3
3 结语
建立了农药乳油制剂中54 种常见农药的气相色谱–质谱筛查方法,该法操作简单,准确度高,精密度好,可以满足杀虫剂中非法添加成分的快速筛查和定量的要求,同时可以参考应用于其它农药制剂或农药类型中非法添加成分的检测。
猜你喜欢
四川蚕业(2022年2期)2022-11-19 02:10:04
预防青少年犯罪研究(2022年1期)2022-08-15 00:35:32
电子技术与软件工程(2019年21期)2020-01-16 05:55:44
时代英语·高一(2019年5期)2019-09-03 02:09:34
深圳职业技术学院学报(2018年3期)2018-07-23 06:42:18
电信科学(2017年6期)2017-07-01 15:44:53
电测与仪表(2016年11期)2016-04-11 12:20:42
湖南农业(2016年4期)2016-03-11 07:03:55
电源技术(2015年5期)2015-08-22 11:18:28
肝胆胰外科杂志(2015年1期)2015-02-27 11:11:30
