
马氏珍珠贝中核苷类成分的分离富集工艺研究
2020-09-23 12:32:34杨书婷韦小翠颜媛媛朱宇超张晓云何瑞欣徐雅蝶程建明
食品工业科技 2020年17期
张 焱,杨书婷,3,韦小翠,颜媛媛,朱宇超,嵇 晶,3, 王 琪,张晓云,何瑞欣,徐雅蝶,程建明,3,*
(1.南京中医药大学药学院,江苏南京 210023;2.江苏省中药功效物质重点实验室,江苏南京 210023;3.江苏省海洋药物研究开发中心,江苏南京 210023)
2015版《中国药典》记载马氏珍珠贝是生产珍珠的珍珠贝品种,并且马氏珍珠贝和三角帆蚌分别是海水与淡水养殖的主要品种,资源十分丰富,但是目前马氏珍珠贝软体在取珠之后弃之不用,造成了大量的资源浪费。经研究,马氏珍珠贝软体中核苷含量为16.23 mg/g(干重),核苷是组成人体结构的基本单位,主要包括碱基、核苷、核苷酸及其衍生物,其具有抗病毒、抗氧化、抗炎、抗肿瘤、免疫调节、醒酒保肝[1-5]等多种重要功能与作用。因此,从马氏珍珠贝软体中分离纯化得到核苷,既能对废弃资源进行废物利用,提高低值贝类的附加值,又可以得到天然核苷。
目前,对核苷类成分的分离纯化技术多种多样。岳明等[6]概括出主要方法有,薄层色谱法、离子交换树脂、硅胶柱层析、大孔吸附树脂、活性炭吸附层析等。对于成分组成复杂的物质,一步分离效果较差,需要多种色谱技术连用才能得到较好分离效果[7-9]。大孔吸附树脂主要利用氢键和范德华力对目标成分吸附[10-11]。并且,大孔树脂具有稳定性高、对目标成分选择性好、操作简单高效等优点[6]。对非离子型核苷类成分具有较好的分离能力。因此,本实验采用大孔树脂技术,首次对马氏珍珠贝上清液中的核苷类成分进行富集与纯化,以期解决马氏珍珠贝软体的资源浪费问题,对其进行二次开发。
1 材料与方法
1.1 材料与仪器
马氏珍珠贝(Pinctadamartensii) 采于广西北海(201807),经南京中医药大学刘圣金副教授鉴定其品种为马氏珍珠贝;腺嘌呤、尿嘧啶、胞苷、次黄嘌呤、黄嘌呤、胸腺嘧啶、2′-脱氧鸟苷、2′-脱氧尿苷、2′-脱氧肌苷、胸苷、尿苷、肌苷、胸苷 均购自美国Sigma公司;HPD100、HPD250、HPD300、HPD400、HPD450、HPD500、HPD600、AB-8、D101大孔树脂 河北沧州宝恩吸附材料科技有限公司;SP207大孔树脂 北京绿百草科技发展有限公司;95%乙醇、甲醇 色谱级,德国默克有限公司。
Waters 2695高效液相色谱仪、Waters 2489 UV检测器 美国Waters公司;DGG-9140型A型电热恒温鼓风干燥箱 上海海森信实验仪器有限公司;FA2004型电子天平 上海天平仪器有限公司;Milli-Q超纯水机 美国Millipore公司;TS-110×50水浴恒温振荡器 上海捷呈实验仪器有限公司;R-220旋转蒸发器 BUCHI Labortechnik AG。
1.2 实验方法
1.2.1 马氏珍珠贝核苷提取 取新鲜的马氏珍珠贝软体,沥干,称重,加3倍量蒸馏水煎煮两次,每次45 min,合并提取液,将合并液减压浓缩至原软体质量的三分之一左右,8000 r/min离心10 min,弃去沉淀,取离心后的上清,加95%乙醇至最终醇浓度为80%,4 ℃静置过夜,抽滤。将上清液70 ℃减压回收,直至提取液无醇味,得到马氏珍珠贝提取液[12-13]。
1.2.2 核苷类成分含量测定
1.2.2.1 标准品溶液制备 精密称定黄嘌呤核苷对照品10 mg,置于50 mL容量瓶中,其余12种核苷对照品精密称定10 mg,置于10 mL容量瓶中,加入15%甲醇定容,摇匀。分别精密吸取鸟苷3.5 mL、胞苷1.0 mL、2′-脱氧鸟苷2 mL、肌苷5 mL、次黄嘌呤15 mL、胸腺嘧啶1.5 mL、尿苷1 mL、2′-脱氧尿苷3 mL、腺嘌呤0.5 mL、2′-脱氧肌苷5 mL、胸苷2 mL于50 mL容量瓶中,15%甲醇定容,得核苷混合对照品。
1.2.2.2 供试品溶液制备 分别精密称取珍珠贝粉0.5 g,加入30倍量水超声提取60 min,8000 r/min离心10 min,取上清,重复提取3次,合并上清液,定容至50 mL容量瓶中,过0.22 μm有机滤膜。
1.2.2.3 色谱条件 色谱柱:Waters X Select HSS C18(250 mm×4.6 mm,5 μm);进样体积5 μL,流动相A为甲醇,流动相B为0.1%磷酸水,流速0.6 mL/min,波长254 nm,梯度洗脱:0~12 min,0.3%~1.0% A,12~20 min,1%~2% A,20~30 min,2%~8% A,30~50 min,8%~15% A。
1.2.2.4 线性关系考察 取“1.2.2.1项”项下的一系列不同浓度的核苷标准品,按照“1.2.2.3项”色谱条件进行HPLC分析。以核苷峰面积为Y,核苷浓度为X做回归计算,得回归方程。
1.2.2.5 精密度考察 使用“1.2.2.2项”方法制备核苷供试品溶液,将同一份供试品在色谱条件下连续进样6次,考察精密度。
1.2.2.6 重复性考察 使用“1.2.2.2项”方法制备6个供试品溶液,根据“1.2.2.3项”色谱条件分析,记录相应的峰面积,计算RSD,对方法重复性进行考察。
1.2.2.7 稳定性考察 将放置在常温条件下的样品,按照“1.2.2.2”供试品溶液制备方法制备一份样品液,分别在0、2、4、8、12、24 h进样,对样品的稳定性进行考察。
1.2.2.8 加样回收率考察 将核苷标准品以100%量加入0.5 g样品中,平行制备六份,进入液相检测,计算加样回收率。
1.2.3 大孔树脂纯化工艺考察
1.2.3.1 树脂型号对核苷吸附率与解吸附率的影响 称取100 g HPD100、HPD250、HPD300、HPD400、HPD450、HPD500、HPD600、AB-8、D101、SP207型大孔树脂,先以95%乙醇浸泡过夜,浸泡过程中每隔2 h轻轻搅拌一次,湿法装柱,用95%乙醇洗脱,洗至流出液加水不浑浊,再使用蒸馏水洗脱至流出液无醇味后,备用。
将10种已处理好的树脂使用滤纸将表面水分吸干后,精密称取5 g(湿重),转移至100 mL具塞锥形瓶内,加入稀释适当倍数的马氏珍珠贝核苷提取液50 mL,放于摇床上在25 ℃水浴中振荡吸附24 h,取出。对吸附后的溶液抽滤,并将得到的滤液放置于50 mL离心管,采用HPLC-UV分析,测定滤液中核苷成分的含量,计算吸附率[6]。
使用滤纸将吸附后的树脂表面溶液吸干,将树脂再次置于新的100 mL锥形瓶内,精密量取体积为50 mL的5%乙醇加入,混匀,于摇床上25 ℃振摇解吸24 h,抽滤并收集滤液,进入HPLC-UV检测解吸液中核苷成分的含量,计算核苷的解吸附率[11]。
吸附率与解吸附率公式如下:Q=(C0-C1)V/M
A=(C0-C1)/C0×100
D=C2/(C0-C1)×100
式中,Q为饱和吸附量(mg/g),A为吸附率(%),D为解吸率(%),C0为吸附前原样品液中总核苷浓度(mg/mL),C1为吸附后所得滤液中总核苷浓度(mg/mL),C2为解吸后滤液中总核苷浓度(mg/mL),V为溶液体积(mL),M为树脂重量(g)[12-13]。
1.2.3.2 上样浓度对吸附效果的影响 马氏珍珠贝水提醇沉上清液经回收乙醇后,对其水部位进行适量浓缩,制备得到不同的核苷浓度(0.27、0.55、1.09、2.19、4.38 mg/mL)作为上样液,以考察上样浓度对SP207树脂吸附效果的影响。
1.2.3.3 上样pH对SP207大孔树脂吸附效果的影响 取核苷浓度为2.19 mg/mL的样品液作为上样液。pH计测得上样溶液(浓度为2.19 mg/mL)pH为4.6。再分别采用适当浓度的盐酸和氢氧化钠溶液将上样溶液调节为pH3.0、6.0、7.0、8.0,以相同流速2 BV/h通过层析柱,至样品液全部通过层析柱后,2 BV水除杂,收集未吸附液与水洗液,量取体积,HPLC测定其中核苷含量,计算吸附率。
1.2.3.4 上样流速对大孔树脂的吸附效果的影响 取核苷浓度为2.19 mg/mL的样品液作为上样液。调节溶液通过层析柱的流速为1、2、3、4、5 BV/h,至样品液全部通过层析柱后2 BV水进行除杂,收集过柱液与水洗除杂部位,量取体积,HPLC测定其中核苷含量,计算吸附率。
1.2.3.5 泄漏曲线考察 量取25 mL预处理好的SP207大孔树脂,湿法填充层析柱(Φ 1.6 cm×40 cm)。调节上样流速为2 BV/h,取300 mL核苷浓度为2.19 mg/mL上样液,以10 mL容量瓶为一流份,样品液全部通过层析柱后,HPLC测定其中核苷浓度,以收集的流份数为横坐标,以流出液中核苷浓度Ct与上样浓度Co的比值为纵坐标,制备泄漏曲线。
1.2.3.6 最佳上样量范围考察 取核苷浓度为2.34 mg/mL的上样液,调节上样流速是2 BV/h,分别加入20、25、30、40、50 mL上样液,待样品全部流过树脂后,用2 BV水除杂,再用 3 BV 10%乙醇以2 BV/h洗脱,收集10%乙醇洗脱液,HPLC分析洗脱液,并通过将洗脱液置于蒸发皿中蒸干以测定该部位固体含量(以下简称“固含”),计算纯化后核苷纯度及转移率。
固体含量=M1-M2
其中:M1=样品重量+蒸发皿重量;M2=蒸发皿重量。
纯度(%)=总核苷含量/固体含量×100
转移率(%)=洗脱液中总核苷含量/上样液中总核苷含量×100
1.2.3.7 水洗量对大孔树脂洗脱效果的影响 样品液全部流过层析柱后,用1、2、3、4、5 BV 蒸馏水以2 BV/h的流速除杂,再以3 BV的10%乙醇以2 BV/h洗脱。收集10%乙醇洗脱部位,使用HPLC分析。并测定10%乙醇洗脱部位的固含,计算核苷的转移率及纯度。
1.2.3.8 洗脱剂浓度对大孔树脂洗脱效果的影响 样品溶液全部流过树脂后,使用2 BV水以2 BV/h进行水洗除杂,再以3 BV水、5%、10%、15%、20%乙醇以2 BV/h洗脱,收集洗脱液。将洗脱液HPLC分析,并测定不同乙醇浓度洗脱部位固含,计算其核苷含量,计算转移率及纯度。
1.2.3.9 洗脱流速对大孔树脂洗脱效果的影响 样品溶液全部流过树脂后,使用2 BV水以2 BV/h进行水洗除杂,再以3 BV 15%醇以1、2、3、4 BV/h洗脱,收集洗脱液。将洗脱液HPLC分析,并测定洗脱液固含,计算洗脱液中核苷含量,计算转移率及纯度。
1.2.3.10 洗脱体积对大孔树脂洗脱效果的影响 样品溶液全部流过树脂后,使用2 BV水以2 BV/h进行水洗除杂,再以3、6、9、12、15 BV 15%醇以3 BV/h洗脱,收集15%乙醇洗脱部位。将15%乙醇洗脱液采用HPLC分析,并测定其洗脱液中固含,计算洗脱液中核苷含量,计算转移率及纯度。
1.2.3.11 径高比对大孔树脂洗脱效果的影响 将SP207树脂分别装入不同规格的层析柱内,使其径高比为1∶1、1∶3、1∶5、1∶7、1∶10。调节溶液流速为2 BV/h,样品溶液全部流过树脂后,使用2 BV水以2 BV/h进行水洗除杂,再以3 BV 15%醇以3 BV/h洗脱,收集15%乙醇洗脱部位。将15%乙醇洗脱液采用HPLC分析,并测定其洗脱液中固含,计算洗脱液中核苷含量,计算转移率及纯度。
1.2.3.12 马氏珍珠贝核苷类成分纯化工艺验证 马氏珍珠贝醇沉上清经过回收乙醇后,其水溶液部位进行浓缩,制备得到核苷浓度为2.19 mg/mL的核苷部位,作为上样液。
分别量取3份处理好的SP207树脂25 mL,湿法装柱(Φ 1.6 cm×40 cm),其径高比为1∶7。调节溶液流速为2 BV/h,将马氏珍珠贝核苷提取液缓慢贴壁加入装有树脂的层析柱中,待样品溶液全部流过树脂后,使用2 BV水以2 BV/h进行水洗除杂,再以3 BV 15%醇以3 BV/h洗脱,收集15%乙醇洗脱部位。将15%乙醇洗脱液采用HPLC分析,并测定其洗脱液中固含,计算洗脱液中核苷含量,计算转移率及纯度。
1.2.4 UV法测定核苷含量
1.2.4.1 肌苷和样品的全波扫描 由于肌苷是马氏珍珠贝中含量高且活性好的成分。因此,选择肌苷作为UV的对照品。
精密称取肌苷标准品10.00 mg,用15%甲醇溶解定容至100 mL容量瓶内,配制成浓度为0.1 mg/mL的肌苷溶液储备液。使用移液管分别量取0.5、0.8、1.0、1.2、1.5、2.0 mL肌苷对照品置于10 mL容量瓶内,用15%甲醇定容,即得系列肌苷标准品溶液,另取1.2.3.12项下马氏珍珠贝核苷纯化液,在200~400 nm进行全波扫描。
1.2.4.2 肌苷标准曲线的绘制 取1.2.4.1种配制的肌苷溶液,进行紫外分光光度计分析,并根据溶液浓度和对应的吸光值绘制标准曲线。
1.2.4.3 马氏珍珠贝纯化液核苷含量测定 取1.2.3.12项下马氏珍珠贝验证工艺中的样品,进紫外分光光度计分析。
1.3 数据处理
2 结果与分析
2.1 核苷类成分含量测定方法学考察
2.1.1 线性关系考察 13种核苷标准曲线、检测限和定量限检测结果见表1,均在线性浓度范围内关系良好,R2>0.999。

表1 13种核苷标准曲线、检测限和定量限检测结果
2.1.2 精密度考察 样品中胸腺嘧啶、尿嘧啶、次黄嘌呤、尿苷、2′-脱氧尿苷、鸟苷、2′-脱氧鸟苷、肌苷、胞苷、2′-脱氧肌苷、腺嘌呤、胸苷、黄嘌呤的精密度RSD分别为0.32%、0.29%、0.24%、0.52%、0.27%、0.23%、0.17%、0.18%、0.45%、0.32%、0.36%、0.36%、0.28%,精密度符合2015版《中国药典》规定,具体结果见表2。

表2 13种核苷精密度实验结果
2.1.3 重复性考察 样品中腺嘌呤、胞苷、黄嘌呤、尿苷、2′-脱氧尿苷、鸟苷、2′-脱氧鸟苷、肌苷、次黄嘌呤、2′-脱氧肌苷、尿嘧啶、胸苷、胸腺嘧啶的重复性RSD分别为1.24%、0.44%、0.71%、1.64%、0.68%、1.57%、2.60%、1.37%、0.67%、0.42%、0.72%、0.47%、0.80%,表明该方法的重复性良好,具体结果见表3。

表3 13种核苷重复性实验结果
2.1.4 稳定性考察 样品中胸腺嘧啶、腺嘌呤、尿嘧啶、胞苷、次黄嘌呤、尿苷、2′-脱氧尿苷、鸟苷、2′-脱氧鸟苷、黄嘌呤、肌苷、2′-脱氧肌苷、胸苷的稳定性RSD分别为0.18%、1.88%、1.33%、0.84%、0.97%、0.41%、1.10%、1.43%、2.86%、1.03%、1.64%、0.67%、0.41%,13种核苷在24 h内稳定,具体结果见表4。

表4 13种核苷稳定性实验结果
2.1.5 加样回收率考察 样品中腺嘌呤、尿嘧啶、胞苷、次黄嘌呤、黄嘌呤、尿苷、胸腺嘧啶、2′-脱氧尿苷、鸟苷、2′-脱氧鸟苷、肌苷、2′-脱氧肌苷、胸苷的平均加样回收率在88.63%~103.33%。RSD值在0.02%~3.91%范围内,表明该测定方法的准确度符合要求,该研究方案科学可靠,具体结果见表5。

表5 13种核苷加样回收率实验结果(n=6)
2.2 大孔树脂纯化工艺考察
2.2.1 树脂型号对核苷吸附率与解吸附率的影响 不同型号大孔树脂对核苷类成分的静态吸附及解吸情况见表6。

表6 不同型号树脂对核苷类成分的静态吸附率及解吸附率
实验筛选了10种树脂,由于树脂吸附原理为相似相容原理,不同型号的大孔树脂极性不同,导致树脂吸附和解吸附性不同。由表6结果可知,在10种大孔树脂中,SP207对核苷的吸附率最高,为63.50%,解吸附率也较好,为73.80%。综合考虑吸附率与解吸附率,最终选择SP207为纯化树脂,对其进行下一步的上样工艺考察。
2.2.2 上样浓度对吸附效果的影响 上样浓度对吸附效果的影响见表7。

表7 不同上样浓度对SP207树脂吸附效果的影响
由表7可以看出,随着核苷上样浓度的增加,吸附率先上升后下降,当上样浓度为2.19 mg/mL达到最大值83.81%。推测可能的原因是当上样浓度较低时,树脂吸附需要较长时间,且比吸附量下降,造成吸附率降低;上样浓度较高时,树脂的吸附推动力大,有利于树脂对目标成分的吸附,吸附率也将增加。但上样液的浓度过高时,有效成分泄漏,吸附效果变差,且导致树脂的再生周期短,因此最终确定SP207大孔树脂最佳上样浓度为2.19 mg/mL。
2.2.3 上样pH对SP207大孔树脂吸附效果的影响 上样pH对SP207大孔树脂吸附效果的影响结果见表8。

表8 上样pH对大孔树脂吸附效果的影响
如表8所示,在SP207树脂吸附过程中,不同的上样pH对树脂吸附核苷有明显的影响。本实验考察了从酸性到碱性的区间,结果显示,随着pH增加,吸附率呈现先增加后趋于稳定最后降低的趋势。表8中显示吸附最佳pH为6(82.78%),但与pH为4.6(不调pH)吸附率(82.14%)相近,因此,考虑工业生产的方便性。选择不调节pH上样,即pH4.6上样。
2.2.4 上样流速对大孔树脂吸附效果的影响 上样流速对大孔树脂吸附效果的影响结果见表9。
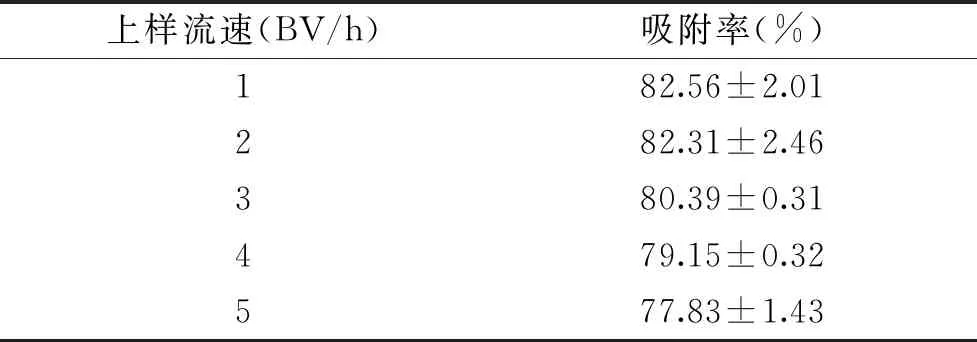
表9 上样流速对大孔树脂吸附效果的影响
如表9所示,随着上样速度的增加,核苷吸附率呈现不断降低。推测可能的原因是上样流速较低时可以使上样溶液缓慢通过树脂,上样溶液与树脂吸附更高效;随着流速的不断增加,上样液与树脂接触不充分,导致成分还未被吸附就随着上样液流出,导致吸附效率降低。由表9可以看出1~2 BV/h的吸附率相近,因此,考虑工业生产效率选择2 BV/h作为上样流速。
2.2.5 泄漏曲线考察 泄露曲线结果见图1。

图1 SP207树脂中核苷类成分的泄漏曲线
由图1显示,随着核苷上样体积的增加,泄漏出现不断增加的趋势,直到树脂达到饱和后不再增加。上样体积为10~60 mL时,核苷类成分呈现缓慢泄漏,上样70 mL时核苷类成分呈现明显泄漏。SP207树脂最大上样量为210 mL,上样10 mL时已经出现泄漏,考虑到吸附效率,需要对数值的最佳上样量进行考察。
2.2.6 考察最佳上样量范围 最佳上样量范围结果见表10。
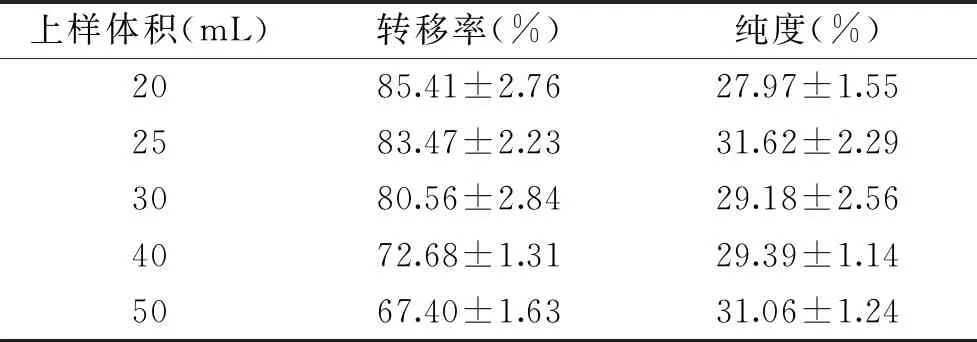
表10 考察最佳上样量范围
表10显示,随着上样体积的增加,核苷转移率呈现逐渐下降趋势,纯度是先上升后下降再上升。分析原因,当上样体积少时核苷树脂对其吸附充分,但杂质吸附量也相应增加,导致转移率高但纯度低;随着上样体积的增加,一定体积的树脂吸附能力逐渐减弱,因此,转移率降低;同时对杂质的吸附能力降低会导致纯度升高。但当随着上样量的继续增加导致核苷泄漏增加,核苷转移率降低,最终同样导致纯度降低。因此,核苷最佳上样体积为25 mL。
2.2.7 水洗量对大孔树脂洗脱效果的影响 水洗量对大孔树脂洗脱效果的影响结果见表11。
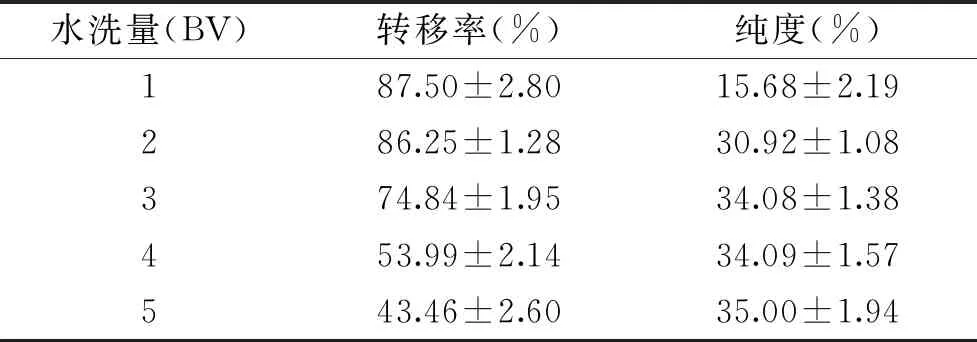
表11 水洗量对 SP207树脂洗脱效果的影响
由表11可知,随着水洗量的增加,核苷转移率逐渐降低,核苷纯度逐渐升高。分析原因,随着水洗体积的增加不仅除去杂质由于核苷为水溶性成分,因此也会洗脱核苷,导致转移率降低,纯度增加。综合考虑核苷转移率及纯度,最终选择2 BV水作为水洗量。
2.2.8 洗脱剂浓度对大孔树脂洗脱效果的影响 洗脱剂浓度对大孔树脂洗脱效果的影响结果见表12。

表12 不同乙醇浓度对核苷类成分的洗脱情况
由表12可知,随着乙醇浓度的增加,核苷转移率逐渐增加,核苷纯度呈现先增加后降低的趋势。分析原因,SP207为中等极性树脂,根据相似相容原理,当洗脱液极性减小时,核苷洗脱量增加导致核苷转移率增加,纯度升高。但随着醇浓度的升高,杂质也逐渐洗脱,导致纯度降低。综合考虑核苷转移率及纯度,最终选择15%乙醇作为洗脱溶剂。
2.2.9 洗脱流速对大孔树脂洗脱效果的影响 洗脱流速对大孔树脂洗脱效果的影响结果见表13。
由表13可知,在SP207树脂的洗脱过程中,通常流速增加,洗脱剂与核苷类成分交换的时间减少,导致交换不充分,从而被洗脱下来的核苷量会随之减少。当洗脱速度为2、3 BV/h时,两者之间的核苷转移率、除杂率与纯度的效果均相近,考虑生产效率,优先选择3 BV/h的洗脱流速。

表13 洗脱流速对SP207树脂洗脱效果的影响
2.2.10 洗脱体积对大孔树脂洗脱效果的影响 洗脱体积对大孔树脂洗脱效果的影响结果见表14。

表14 洗脱体积对SP207树脂洗脱效果的影响
表14显示,随着洗脱体积的增加,核苷转移率时逐渐增高,纯度呈现先增后降低的趋势。分析原因,随着洗脱体积的增加,可以使核苷类成分洗脱更加充分,但杂质也会被洗脱。因此,核苷转移率会增加,但当核苷全部洗脱后,再继续增加洗脱体积杂质增加导致核苷纯度降低。由表14可以看出,当3 BV洗脱时大部分核苷已经被洗脱,随洗脱体积增加转移率增加不明显。因此,从转移率、纯度以及工业成本、效率等因素。最终,选择3 BV为核苷洗脱体积。
2.2.11 径高比对大孔树脂洗脱效果的影响 径高比对大孔树脂洗脱效果的影响结果见表15。

表15 径高比对SP207树脂洗脱效果的影响
由表15可知,随着径高比的增加核苷转移率与纯度均呈现先增高后降低的趋势,且在径高比为1∶7时达到最高,此时核苷转移率为89.00%,纯度为33.52%。考虑核苷转移率及纯度等因素,最终选择转移率与纯度均最高的1∶7为其径高比。
2.2.12 马氏珍珠贝核苷类成分纯化工艺验证 采用以上筛选出的工艺条件,对马氏珍珠贝进行纯化,纯化结果见表16。
由表16可知,马氏珍珠贝水提醇沉上清液经回收乙醇后经过SP207大孔树脂纯化核苷类成分,其核苷转移率处于较高水平,总转移率为89.49%。核苷类成分纯度从1.81%提高到36.10%。由于HPLC测定的仅仅是核苷与碱基,不包含核苷酸类成分,为了更加全面地测定核苷类成分,本实验将采用UV-Vis法对核苷类成分进行测定。

表16 马氏珍珠贝核苷类成分纯化工艺验证
2.3 UV法测定核苷含量
2.3.1 肌苷和样品的全波扫描 全波长扫描结果显示,252 nm为样品及对照品最大吸收波长,结果见图2。

图2 肌苷和样品全波扫描结果
2.3.2 肌苷标准曲线的绘制 取1.2.4中配制的肌苷溶液,进紫外分光光度计分析,结果图3。

图3 肌苷标准曲线图
2.3.3 马氏珍珠贝纯化液核苷含量测定 对马氏珍珠贝验证工艺中的核苷含量进行测定,结果显示UV测的纯度为58.19%,结果见表17。由于UV检测到的成分中包含了HPLC中未能检测到的核苷类成分,因此含量检测结果更高。

表17 UV测定核苷含量结果
3 结论
本文主要研究马氏珍珠贝纯化工艺,以马氏珍珠贝水提醇沉液中的核苷类成分为指标,采用HPLC测定含量。对大孔树脂型号、上样工艺、洗脱工艺进行考察,富集得到纯度为36.10%的核苷部位,总转移率89.49%。在贝类软体的纯化过程中,极有可能存在其他核苷类成分,为全面测出总核苷类成分纯度,故进一步对核苷纯化部位进行UV测定,结果显示经SP207纯化后的核苷纯度为58.19%。此结果说明,此大孔树脂纯化方法适用于马氏珍珠贝中核苷类成分的分离富集,为马氏珍珠贝的后续开发奠定了基础,同时也为其他同类成分的富集分离提供了分离纯化工艺参考。
猜你喜欢
食品与发酵工业(2024年4期)2024-03-01 12:56:12
数学物理学报(2021年3期)2021-07-19 06:02:44
数学物理学报(2020年6期)2021-01-14 01:00:44
数学年刊A辑(中文版)(2020年1期)2020-05-19 00:30:50
数学物理学报(2018年2期)2018-05-14 07:32:07
中成药(2017年5期)2017-06-13 13:01:12
中成药(2017年5期)2017-06-13 13:01:12
中国当代医药(2015年9期)2015-03-01 02:02:08
肝博士(2015年2期)2015-02-27 10:49:43
中国药业(2014年22期)2014-05-02 08:24:30
