
高效液相色谱-串联质谱测定大鼠脑组织中D -丝氨酸和L-丝氨酸的含量
2020-09-17 04:00陈荣祥
分析科学学报 2020年4期
王 颖, 张 勇, 何 婷, 陈荣祥*
(1.遵义医科大学医学与生物学研究中心,贵州遵义 563000;2.遵义医科大学药学院,贵州遵义 563000;3.遵义市理化分析测试工程技术研究中心,贵州遵义 563000)
氨基酸类化合物广泛存在于生物体内,它们对于调节机体的细胞代谢,疾病预防,疼痛调节等生理活动具有重要作用。大部分氨基酸有D -型和L-型两种对映体,其中L-氨基酸是构成天然蛋白质的底物和生物体内重要的能源构成成分。D -型氨基酸在动物体内也广泛存在并具有不同于L-氨基酸的生理功能[1 - 3]。其中D -丝氨酸(D -serine,D -Ser)是中枢神经系统中N-甲基-D -天冬氨酸受体的信号分子,由丝氨酸消旋酶对L-Ser的对映体转化而来,D -Ser的浓度与许多中枢神经系统疾病尤其是疼痛密切相关。如降低大鼠头端前皮层内源性D -Ser浓度可减轻疼痛相关的负性情绪[4],在创伤及外周炎症刺激下,星形胶质细胞被激活并释放大量D -Ser会进一步导致促痛效果[5,6]。目前,基于丝氨酸消旋酶活性和表达的抑制或增强而引起的内源性D -Ser水平的变化是目前神经科学领域研究的热点,因此,研究并建立D -Ser的准确高效的测定方法对神经科学、临床医学、疾病诊断及控制等具有重要意义。
生物样品基质复杂且氨基酸大多不具有紫外或荧光信号,常需借助衍生化技术将其衍生后再进行分离检测[7]。常用的有高效液相色谱法[8]、毛细管电泳法[9]等。但这些方法普遍存在分析时间长、操作繁琐以及肽和胺类物质的干扰等缺点。近年来,高效液相色谱-串联质谱法(HPLC-MS/MS)由于其高灵敏度、选择性好、低检测限等优点,已被广泛应用于复杂基质中痕量组分的分析测定。采用HPLC-MS/MS测定D -氨基酸,可以直接采用手性色谱柱进行分离[10,11],或者采用手性衍生试剂衍生化后分离[12]。采用衍生化的方式,不仅有利于手性异构体的分离,而且可以提高离子化效率,增加其在反向色谱柱中的保留,消除内源性干扰[13]。用于D -,L-氨基酸分离的衍生化试剂有(S)-(+)-4-(N,N-二甲氨基磺酰)-7-(3-异硫氰酸基吡咯烷-1-基)-2,1,3-苯并恶二唑[14]、Marfey试剂[15]、(S)-NIFE[12]等。但这些衍生化方法普遍存在试剂昂贵,衍生步骤繁琐等缺点。
邻苯二甲醛(OPA)结合N-乙酰-L-半胱氨酸(NAC)是液相色谱中,常用于荧光检测的手性衍生试剂,其衍生方法简单快速。然而生物体内氨基酸种类繁多,采用液相色谱分离干扰严重,分离困难。串联质谱在多重反应监测(MRM)模式下可以通过两次离子选择,有效排除其他氨基酸干扰,其灵敏度、准确度均优于光学检测器。本文利用该衍生试剂对D -Ser、L-Ser进行柱前衍生化,采用高效液相色谱分离,串联质谱法检测,建立了快速灵敏的D -Ser、L-Ser检测方法,并用于痛觉模型大鼠不同脑区D -Ser、L-Ser的含量的测定。
1 实验部分
1.1 仪器与试剂
I-class-TQ-S超高效液相色谱-三重四极杆质谱仪(美国,Waters);ME-104电子天平(梅特勒-托利多);FE-28 pH计(梅特勒-托利多);SB-800DTD型超声清洗仪(宁波新芝生物科技股份有限公司);Microfuge-20型超高速离心机(贝克曼库尔特商贸(中国)有限公司)。
D -Ser、L-Ser、L-刀豆氨酸(L-CAN)、OPA、NAC,均购自上海阿拉丁生化科技股份有限公司,纯度大于98%。甲醇为质谱纯,购于霍尼韦尔贸易(上海)有限公司。NH4Ac为质谱纯,硼砂为分析纯,均购于上海阿拉丁生化科技股份有限公司。实验用水由纯水超纯水系统(Purelab Chorus 2,英国埃尔格)制备。
1.2 标准品储备液和衍生溶液的配制
标准品储备液的配制:分别准确称取D -Ser、L-Ser、L-CAN标准品10 mg,用水溶解并定容至10 mL,制得浓度为1 mg/mL的储备液。其中L-CAN为内标。
衍生溶液的配制:分别称取OPA 15 mg和NAC 100 mg,溶于1 mL甲醇,加入4 mL 0.1 mol/L硼砂缓冲液(pH=10.0)混合均匀,4 ℃储存备用。使用前加0.1 mol/L硼砂缓冲液(pH=10.0)稀释至所需浓度作为工作液。
1.3 实验方法
1.3.1 动物实验成年SD大鼠12只,雄性,体重220~250 g,购于长沙市天勤生物技术有限公司(合格证号:SCXK(湘)2014-0011)。所有大鼠在安静、温度24±1 ℃、湿度60±5%、12 h明/12 h暗光照环境中饲养。首先建立双侧坐骨神经结扎(CCI)痛觉模型,12只大鼠随机分为:A:对照组,B:CCI+生理盐水组。实验开始时,CCI组大鼠于右后肢纵向切开皮肤,钝性分离,暴露坐骨神经主干,丝线间断结扎,间距1 mm,松紧度以小腿肌肉出现轻微震颤为标准,生理盐水冲洗后逐层缝合,术后予腹腔注射1 mL生理盐水(NS)。
1.3.2 样品处理和衍生化反应用异氟烷麻醉大鼠后立即断头处死,迅速取出脑组织并在冰上分离出额叶、顶叶、颞叶、海马、纹状体,按照每0.1 g脑组织加入1.5 mL的HCl(0.02 moL/L),冰浴下匀浆,12 000 r/min离心10 min,取上清液,按照体积比1∶3 加入乙腈沉淀蛋白。12 000 r/min离心10 min,取90 μL离心后的上清液,加10 μL内标(10 μg/mL L-CAN),20 μL衍生试剂和1 080 μL水,涡旋混合反应3 min后进样。衍生化反应如图1所示。
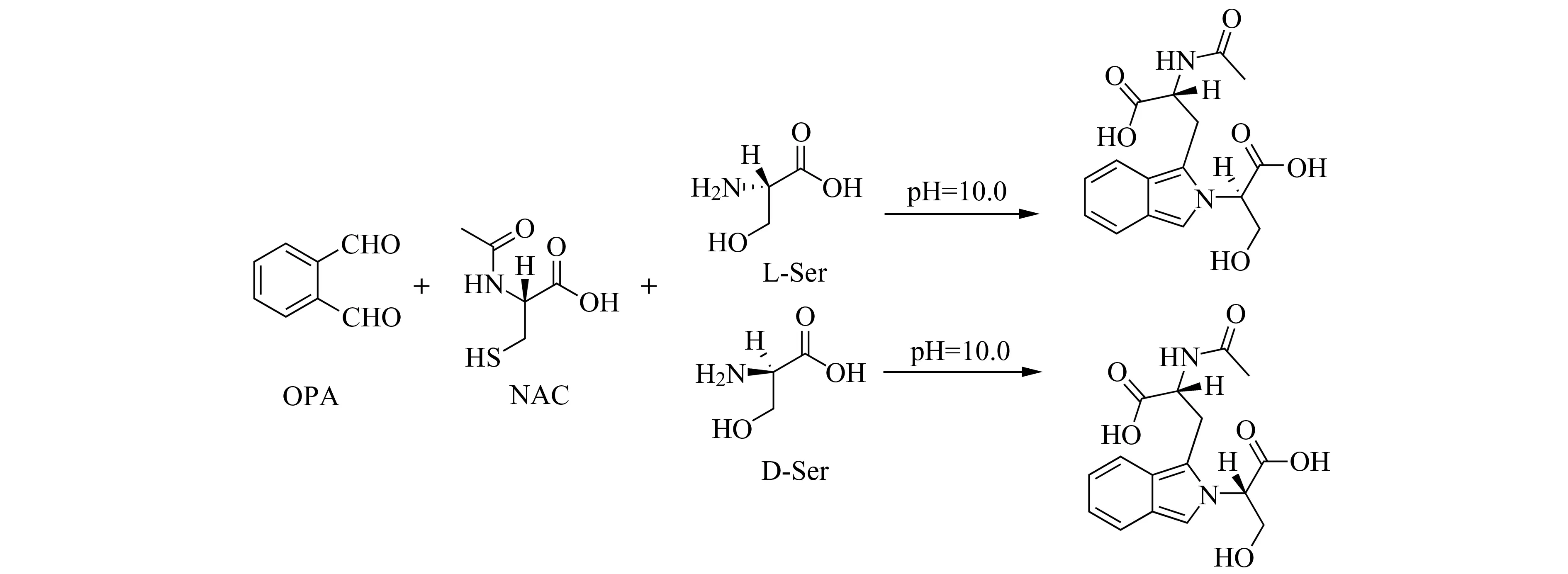
图1 D -Ser、L-Ser与OPA的衍生化反应Fig.1 Derivative reaction of D -Ser,L-Ser with OPA
1.4 色谱和质谱条件
色谱柱:Waters XBridge BEH C18柱(150 mm×3 mm,2.5 μm);流动相:13 mmol/L NH4Ac溶液(A)-甲醇(B)。梯度洗脱:0~5 min,5%~20%B;5~8 min,20%~42.5%B;8~10 min,42.5%~90%B;10~11 min,90%B;11~11.5 min,90%~5%B;11.5~15 min,5%B。体积流量:0.4 mL/min;柱温:40 ℃;自动进样器温度为4 ℃;进样量:5 μL。
电喷雾电离源,采用正离子模式(ESI+);扫描方式为MRM;毛细管电压:3.5 kV;温度:450 ℃;气流量:750 L/h。
2 结果与讨论
2.1 衍生条件的优化
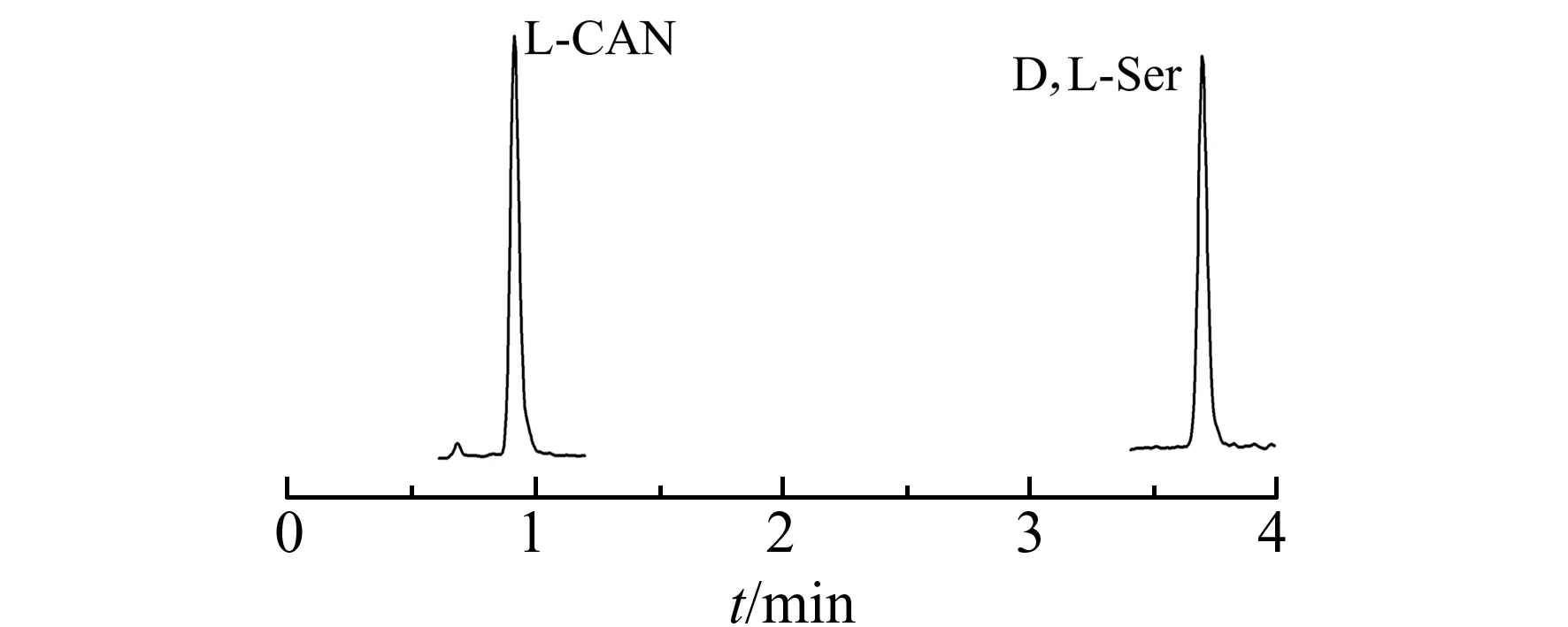
图2 衍生化前色谱图(HILIC色谱柱)Fig.2 Chromatogram of D,L-Ser without derivatization(HILIC column)
采用液相色谱法分离D -Ser和L-Ser存在诸多困难。首先,氨基酸极性很强,在反相色谱柱中难以保留,通常需要在流动相中添加离子对试剂,但这会严重影响质谱性能,降低其负离子模式下的响应。采用HILIC色谱柱可以增加氨基酸的保留时间,但是D -Ser和L-Ser保留时间一致,无法分离,如图2所示。目前拆分D -Ser和L-Ser需要采用手性色谱柱或者手性衍生试剂进行衍生化。例如,Kohnosuke Kinoshita等采用CROWNPAK CR(+)色谱柱,在流动相中添加三氟乙酸分离D -Ser和L-Ser[11]。但是该方法D -Ser和L-Ser的容量因子较低,基质效应严重,需要采用昂贵的同位素内标进行定量。

图3 衍生试剂OPA浓度的优化Fig.3 Optimization of the concentration of OPA
OPA被广泛应用于氨基酸对映体的光学拆分,在碱性环境中,在手性硫醇化合物如NAC或N-叔氧羰基-L-半胱氨酸存在下,OPA容易与伯胺发生反应,生成非对映异吲哚衍生物[16]。一般选择衍生试剂的pH值为9.5~10.5,室温下混合反应3 min即可反应完全[17]。实验中,取目标脑区样品按照上述“1.3.2”项的方法对样品进行前处理,取同等体积样品分别加入浓度为0.002、0.004、0.01、0.02、0.04、0.06、0.1、0.2、0.4 mg/mL的衍生试剂并在室温下反应3 min,过滤后进样,根据峰面积的变化来选择最佳的衍生试剂浓度(图3)。结果显示当衍生试剂的浓度为0.06 mg/mL时衍生产物峰面积已到最大,继续增加衍生试剂的用量峰面积保持稳定。为保证衍生反应完全进行,选择衍生试剂的浓度为0.1 mg/mL。
2.2 质谱条件的优化
一般情况下,含氮化合物在正离子模式下信号较强,因此我们采用了正离子模式监测。通过全扫描模式获得氨基酸衍生物的母离子,在选择离子检测模式下优化其锥孔电压。继续在产物离子扫描模式下获得产物离子,响应强的产物离子作为该物质的定量离子,在MRM模式下优化了碰撞能量。3种目标氨基酸衍生产物的质谱检测参数见表1,D -Ser、L-Ser和L-CAN衍生产物的二级质谱谱图见图4。

表1 目标氨基酸衍生产物的质谱分析参数
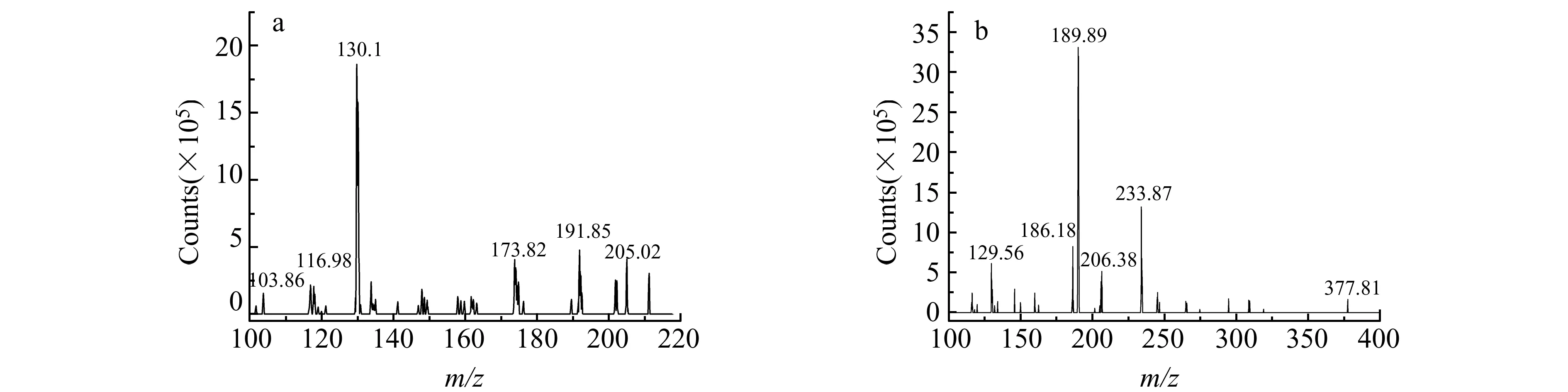
图4 D -Ser、L-Ser(a)和L-CAN(b)的二级质谱图Fig.4 MS2 of D -Ser,L-Ser(a)and L-CAN(b)
2.3 方法学考察
2.3.1 线性关系和检测限配制浓度为0.01、0.02、0.05、0.1、0.2、0.5、1.0、2.0 μg/mL的D -Ser和L-Ser 系列浓度标准溶液,加入等体积OPA-NAC衍生化试剂,充分反应,进样检测,记录色谱图。以标准品与内标物(L-CAN)的浓度之比为横坐标(x),以标准品与内标物的峰面积之比为纵坐标(y)作回归曲线,分别得D -Ser和L-Ser异构体衍生物的回归方程为:y=0.1745x-0.0027(R2=0.9997),y=0.1461x-0.0166(R2=0.9996)。结果表明D -Ser、L-Ser的检测浓度在0.01~2 μg/mL范围内具有良好的线性关系。以3倍信噪比计算检测限,D -Ser、L-Ser的检测限分别为0.4 ng/mL和0.5 ng/mL。而采用非衍生化的方式,D,L-Ser的检测限均为15 ng/mL,可见衍生化不仅有利于二者的分离,而且检测灵敏度也得到提高。
2.3.2 稳定性OPA衍生方法的一个重要缺陷是衍生产物不稳定,以往的研究表明在低温下可以增加产物的稳定性[18]。将进样器的温度设置为4 ℃,在0.1~12 h的范围内以峰面积评定产物的稳定性,结果表明在4 ℃条件下,产物在12 h内稳定。
2.3.3 回收率分别添加不同浓度水平的D -Ser、L-Ser标准品至脑组织提取液中进行加标回收实验,每个浓度5个样品,计算方法回收率,结果见表2。D -Ser、L-Ser平均加样回收率分别87.63%~94.00%和90.93%~99.56%,相对标准偏差(RSD)小于7.82%。
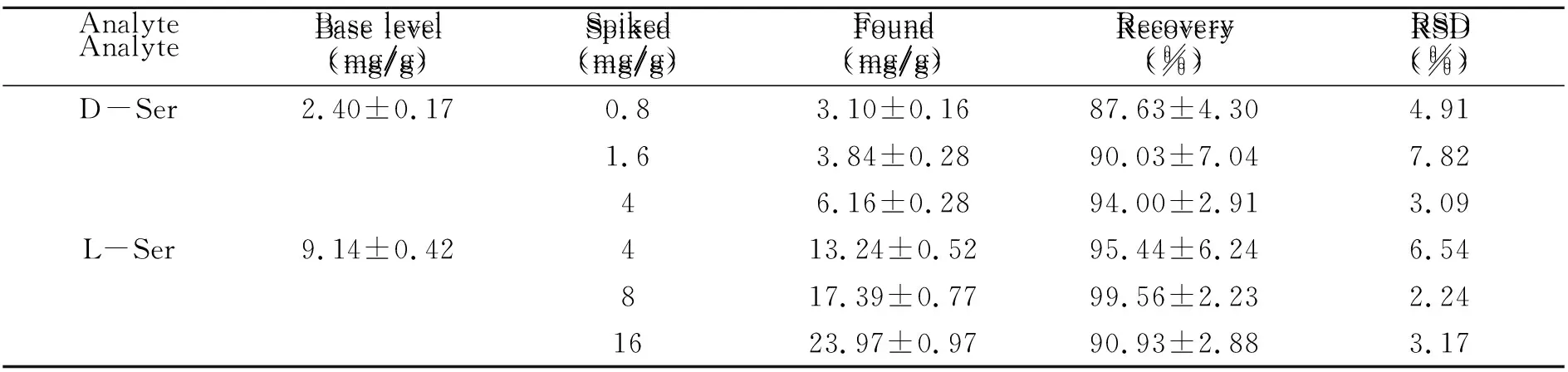
表2 脑组织中D -Ser和L-Ser的回收率(n=5)
2.3.4 精密度取脑组织提取液衍生化后进样测定,连续进样6次,记录检测出的D -Ser、L-Ser的衍生物峰面积值,计算得峰面积值的RSD分别为1.60%、1.30%。结果表明仪器的精密度良好。
2.3.5 基质效应由于D -Ser、L-Ser在动物体内普遍存在,缺乏空白基质,因此我们通过比较在大鼠脑组织提取液中加入标准溶液绘制标准曲线获得的斜率和在标准溶液中获得的斜率来确定基质效应。基质效应(%)=大鼠脑组织提取液中的平均斜率/标准溶液中的平均斜率×100%。本实验中,D -Ser、L-Ser、L-CAN的基质效应分别为106.11%、109.34%、110.20%,基质效应不显著。
2.4 方法学应用
应用上述方法对12只成年大鼠不同脑区进行处理后测定,D -Ser的保留时间为4.66 min,L-Ser的保留时间为4.83 min,供试样品溶液中被测组分的色谱峰达到较好分离,结果见图5。采用上述方法对对照组和疼痛模型组大鼠不同脑区中D -Ser、L-Ser的含量进行了测定,结果见表3。结果表明,疼痛模型组和对照组比较,在额叶、顶叶、海马等脑区D -Ser、L-Ser的含量无明显差异,在颞叶中D -Ser和L-Ser的含量均有显著性的下降(p<0.01)。在纹状体内D -Ser和L-Ser含量增加(p<0.05)。疼痛组和对照组颞叶、纹状体中D -Ser与L-Ser浓度的比值存在显著差异(p<0.05)。因而,脑组织中D -Ser与L-Ser浓度有可能作为神经系统疾病的诊断指标。
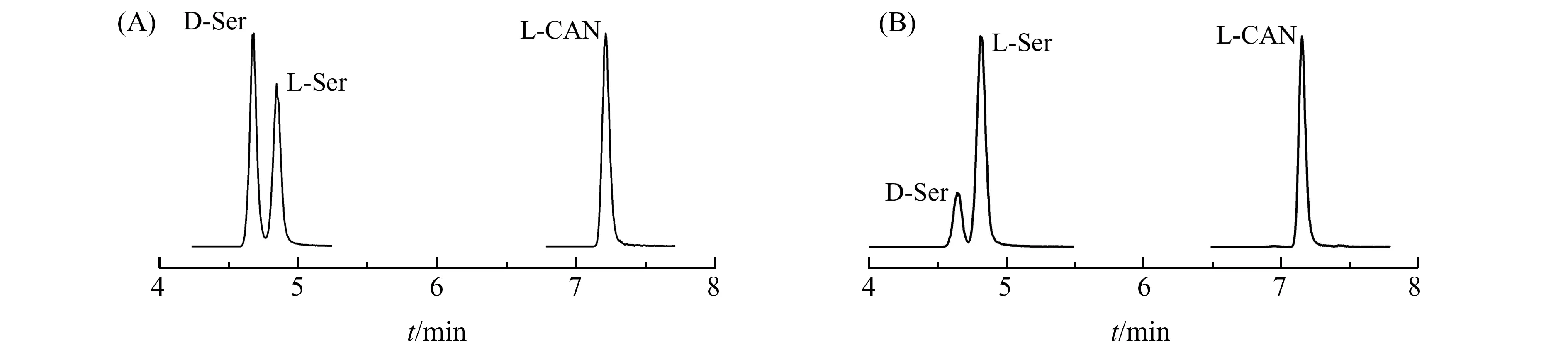
图5 3种氨基酸标准品(A)和生物样品(B)色谱图Fig.5 Chromatograms of the standard solution (A) and brain tissues sample (B)
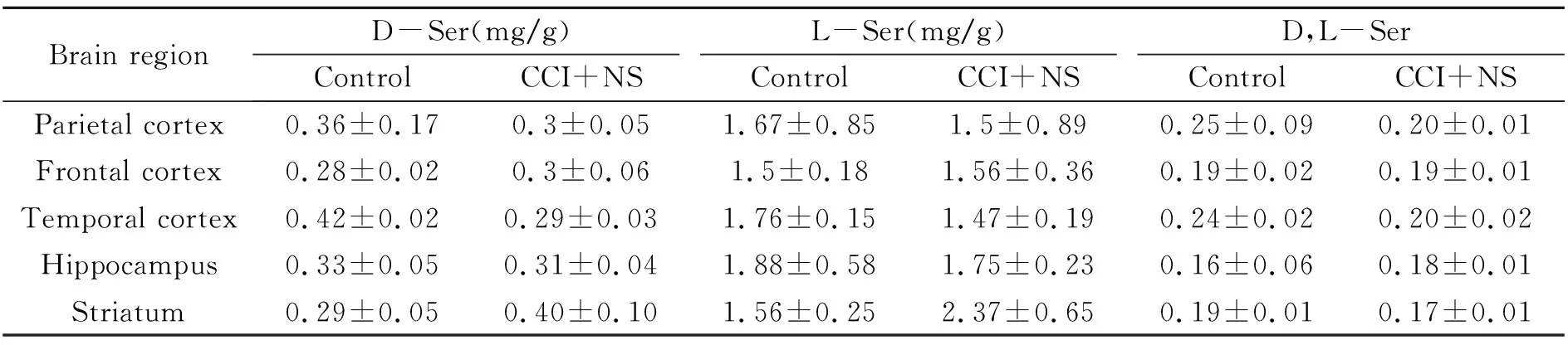
表3 不同脑区D -Ser、L-Ser浓度(n=6)
3 结论
本研究建立了OPA柱前衍生化,HPLC-MS/MS法测定大鼠脑组织中D -Ser、L-Ser的方法,该方法灵敏、快速、专属性好,检测时间短。经方法学验证该方法精密度、稳定性、线性关系、加样回收率均良好,适用于脑组织中D -,L-Ser快速同时检测,对于疼痛相关神经系统疾病的进一步研究提供方法学参考。
猜你喜欢
当代水产(2022年4期)2022-06-05
口腔护理用品工业(2021年4期)2021-11-02
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
现代仪器与医疗(2021年1期)2021-06-09
中国资源综合利用(2017年4期)2018-01-22
现代检验医学杂志(2016年1期)2016-11-12
当代化工研究(2016年5期)2016-03-20
特产研究(2014年4期)2014-04-10
