
The Paradigm of T Cells in Shaping Tumor Microenvironment
2020-09-10 07:22:44DiaRoySayantanBoseSaikatDuttaGaurisankarSa
Trends in Oncology 2020年2期
Dia Roy Sayantan Bose Saikat Dutta Gaurisankar Sa



Abstract
The infiltration of immune cells in the tumor micro-environment is well-documented in cancer patients and the resultant complex interactions are determinants of disease prognosis. Consequently, a proper understanding of this interplay is essential for the development and advancement of therapeutic strategies as well as novel prognostic markers. The co-existence of immune cells with the tumor is often accompanied by an impaired immune response that creates a tumor-promoting micro-environment. T-cell mediated immunity forms the major branch of the immune system that is required to mount an effective response against nascent tumors. Major research in the last few decades indicates that a potent source of immunosuppressive cellular and molecular networks prevailing at the site of tumor is mediated by dysfunctional and defective responses mediated by T cell thereby redirecting and reshaping the destiny of T cell and promoting tumor progression. In this review, we aim to summarize the breakthrough advances in recent years that help us gain a better understanding of the immunosuppressive networks resulting from T-cell anergy, exhaustion, senescence and presence of Treg cells in the tumor micro-environment. We also focus on recent discoveries regarding advance in the Th17 balance, polyfunctionality of T cells as well as T cell stemness that improve our perception of the tumor-immunity interactions. We try to emphasize how such information has an impact on therapeutic development and the clinical outcome of the patients.
Keywords: T cell; Tumor infiltrating lymphocytes; Tumor microenvironment; Treg; CART cell
1. Introduction
Tumor development encompasses numerous changes in genetic level. These alterations further pass into the descendants of the transformed cell over the period of time which ultimately results in the development of a malignant phenotype mainly characterized by uncontrolled proliferation and growth[1].Concomitantly, the surrounding normal tissues encounter series of alterations that leads to the establishment of tumor microenvironment. In the initial stage, these alterations in response to tumor development resemble chronic inflammation-a host response encompassing a multifactorial network of chemical signals mainly designed to “heal” the affected area[1,2]. In fact, Dworak in 1986 indeed described tumor as wounds that do not heal[3].In context of tumor microenvironment, this process of chronic inflammation involves activation and directed migration of immune cells from the venous system to sites of damage. Thus, the role of inflammation in shaping the tumor microenvironment has been widely recognized and referred to as the “host reaction” to the tumor[4]. However, the presence of immune cells in the pre-cancerous or benign tissue as noted by Kornstein et al.and Von Kleist et al. pointed to the fact that tumor appears to be capable of inducing an inflammatory response at an early stage[5]. This has been described as an effort of the host immune system to interfere with tumor growth and progression, alias stated as the “immune surveillance” or “elimination”-one of the 3 E’s of “Tumor Immunoediting”[6]. However, the developing tumor does not accept this without resistance. Instead the tumor cells enter into an “equilibrium stage” in which the immune system controls net tumor cell outgrowth mainly by constraining the growth of the sensitive tumor cells and replacing them with the immunologically resistant tumor cells (immune selection)[6,7]. Finally, this functional sluggishness of the tumor cells gets ruptured that leads to the progression of the tumor cells into the “escape stage”, during which it takes advantage of the host response in order to utilize the host as a contributor in creating the microenvironment suitable for tumor progression in an immunologically unrestrained manner and eventually become clinically detectable ("immune evasion")[6,8]. Thus, the 3E’s of the dynamic “Tumor Immunoediting” process encompasses three series of events- elimination, equilibrium and escape which ultimately decides the fate of the tumor. As proposed by Hanahan and Weinberg, escape from immune control is now acknowledged as one of the major hallmarks of cancer[9]. Thus, the coexistence of immune cell with tumor cells is often corroborated with cancer progression thereby revealing that this coexisting scenario indicates a dysfunctional and compromised immune phenotype[7]. In actual fact, it is well confirmable that a rich provenance of the networks of immunosuppressive molecules in the tumor microenvironment nurture dysfunctional and impaired T cell responses leading to the ultimate redirection of T cell fate and subsequent clinical outcome of the patient. In the present review, we tried to summarize the role of T cells in tumor fate determination and also the acquired dysfunctions of effector T cells, as mediated by other T cells in context of tumor microenvironment. In addition, we tried to shed light in the advance of Th17 equilibrium, the stemness property of T cell and also the multifunctional attribute of T cells which altogether may result in a better clinical outcome of the patients.
2.Types of Immune Cells in Tumor Microenvironment
Immune responses to malignant cells are categorized mainly as locoregional or systemic response[5,10]. Locoregional responses mainly encompass the tumor infiltrating lymphocytes (TILs) which accumulate in the tumor microenvironment (in case of solid tumors). TILs isolated from numerous human tumors are functionally different and somewhat compromised than their normal counterparts. On the other hand, the systemic immune response as measured in the peripheral circulation of cancer patients is particularly elusive. Recent data have shown that the components of systemic immune response show similar functional impairment like the TILs. However, this does not mean that cancer patients are immunodeficient.
The anti-bacterial or anti-viral responses of the patients remain unaltered but the tumor-specific immunity gets compromised. Numerous studies have shown that human tumors are mainly infiltrated by T cells (CD3+TCR+) which comprises the largest component of mononuclear tumor infiltrates in human tumors (Mihm et al., 1996). This is followed by other immune cells namely macrophages (CD45+CDI4+ cells) referred to as TAMs (Tumor associated macrophages), dendritic cells and B cells [11,12]. Apart from this, human tumors are sometimes permeated by granulocytes. By far the most frequent cell in tumors has characteristics of immature myeloid cell (iMC) expressing CD33 as common myeloid marker [13], but deficient of mature lymphoid cell markers. In tumors, under oxygen deficient hypoxic condition, iMC might contribute to creating conditions favoring T-cell suppression by generation of H2O2 [14].
3. Immune-Suppressive Networks in Tumors
Over time, the phenotype as well as the function of effector T cells in context of tumor immunity has been an active area of research. Studies have been made in different types of murine models that enable us to focus on the role of immune system in tumor progression. Tumor microenvironment is endowed with a well-formed immunosuppressive network which helps in the initiation and maintenance of the dysfunctionality of T cells[11]. Pertinent T cell activation depends on the interaction of TCR-specific stimulatory antigen presented with Major Histocompatibility molecules (MHC) present on surface of the Antigen presenting cell (APC) and association of CD28 with members of B7 stimulatory family (CD80 and CD86)[11,12]. This co-engagement triggers the intracellular signaling via transcription factors like nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and Nuclear Factor of Activated T cells (NFAT) that ultimately promotes activation of T cells. On the other hand, T cell inactivation is of equal importance for the immune reaction to be dampened and for the establishment of homeostasis. Numerous immunosuppressive Antigen Presenting cells and other immune cells like regulatory T cells facilitate this process by helping the effector T cells to enter a dormant phase. Following activation, T cells also start expressing numerous co-inhibitory receptors like PD-1(Programmed Death-1) and CTLA-4 or CD152(Cluster of Differentiation) which helps in suppression thereby resolving immune responses. In addition, expression of other negative regulators of T-cell functionality like Tim-3, KLRG-1, PD-1, LAG-3, CD57and CD160 also helps to facilitate T cell unresponsiveness[13] (Figure 1). Tumor malignancies develop certain machineries by which they can seize these immunosuppressive mechanisms thereby maintaining a potent and vigorous state of suppression[14]. After getting a brief overview about the immunosuppressive factors that contribute to T cell dysfunctionality, we will try to take a deeper look into the fate of T cells in tumor microenvironment and how it shapes the overall tumor microenvironment.
4. Role of T Cell in the Immunological Sculpting of Tumor Microenvironment
CD3+TCR+ T cells comes out to be an important constituting factor amidst several cells present at the tumor microenvironment. Although in few cases as shown by Lin et al. macrophages can profusely infiltrate the tumor sites, but in most human tumors, T cells constitute the largest magnitude of the tumor infiltrating components (Mihm et al,1996)[15]. Release of sufficient number of immunogenic antigens during the initial stage of tumor development results in the priming of the naïve resting T cells at the draining lymph node followed by their activation and consequent migration to the tumor site. This results in the elicitation of an effective and potent anti-tumor immunity resulting in the elimination of the immunologically sensitive tumor cells[7,16]. Histopathological studies of tumor mass reveal the presence of tumor-associated T cells beyond the invasive periphery of the tumor extending till the hypoxic core of tumor[17]. Nevertheless, it has been shown that the anti-tumoral property of these T cells is rather auxiliary and indirect because these cells mainly contributes to cancer immunosurveillance following tumor abscission[15]. Proposition stating that anti-tumor cytolytic T cells (mainly CD8+T cells) constitute a proportion of Tumor infiltrating T-lymphocytes(TIL-T) has been well acknowledged however early limiting dilution studies conducted with TIL-T obtained from many human tumors suggested that the prevalence of such cytotoxic T lymphocytes was low as compared to that obtained from the peripheral blood T lymphocytes[18]. The antigen presenting cell (APC) primes and activates the CD8+ T cells which subsequently differentiates into the CTLs and exercise an effective anti-tumoral effect through the release of perforin and granzyme containing granules Hanson et al. 2000; Matsushita et al. 2012)[19]. Alongside, CD4+ T helper cells(Th1) also mounts anti-tumor response via secretion of numerous proinflammatory cytokines like IL-2,TNF-α, and IFN-γ which not only promotes the priming of T cells and CTL cytotoxicity but also mediate the anti-tumoral properties of macrophages and NK cells (Kalams and Walker 1998; Pardoll and Topalian 1998; Shankaran et al. 2001)[20]. Concomitant presence of tumor infiltrating CD8+ T cells as well as Th1 cytokines in tumor microenvironment shows a positive correlation with a favorable prognosis in terms of both overall survival as well as recurrence free survival (RFS) in numerous human tumors. Additionally, Vβ-restricted clones of T cells existing in isolated TIL under some condition, can recognize and selectively kill autologous tumor cells thereby promoting anti-tumor effect (Weidmann et al., 1992). Now the question arises if the T cells are so potent at killing the malignant cancerous cells, then how do some tumor cells manage to escape this effective immunosurveillance mechanism as devised by the effector T cells (Figure2). Experimentations have shown that tumor cells maneuver the immunosuppressive properties of T cells and also disrupt the effector function of the anti-tumor cytotoxic T cells (Grivennikov et al. 2010)[21]. As described previously, the effectiveness of effector T cells to mount an anti-tumor response depends on their ability to recognize the tumor antigen(immunogenic) and the presence or absence of co-inhibitory signals which can impair the function of T cells (Speiser et al. 2016)[22]. Consistently, it is widely concurred that in a T-cell dependent procedure, most malignant cells expressing immunogenic epitopes will be recognized and subsequently destroyed during the initial stages of cancer development (Matsushita et al. 2012)[23]. However a small pool of less immunogenic tumor cells manage to escape this immunosurveillance machinery of T cells and thereby survive- a process termed as immune escape[7]. It finally resulted in the survival of only those cancer cells that can adopt an immune-resistant phenotype. Concomitantly, tumor cells devise certain mechanisms that can mimic the peripheral tolerance thereby preventing the cytotoxic effect of effector T cells and other anti-tumor immune cells like NK cells, macrophages etc(Figure 2). Since T cells play a major role in shaping the tumor landscape, we will now try to unravel the role of two major CD4+T cell subtype-Treg cell and Th17 cells in shaping the tumor landscape.
4.1 Regulatory T cells: Inducer of Dysfunctionality of T Effector Cells in Tumor Milieu
(a) Discovery and Steady Characterization of Regulatory T cells (Treg)
Recent researches on tumor immunology highlights the role of regulatory T cells (Treg) in tumor progression. Sakaguchi and his colleagues for the first identified this cell as CD4+ CD25+ T cells in the year of 1995[24]. Though the presence of CD25 is adequate to isolate a uniform population of nTregs in murine models but the same perspective appeared to be rather difficult when utilized for the isolation of human Treg cells. In human, expression of CD25 is not exclusive for Treg cells, rather its presence can be observed at varying levels of around 35% in total human T cell. As shown by Baecher Allan et al., in 2004, T effector cells profusely express CD25 upon receiving the stimulatory signals[25]. Continual exposure of humans to numerous immunogenic challenges unlike the murine system which is kept under pathogen-free conditions, results in T cell activation in human which is the main cause for CD25 upregulation in human effector T cells. Finally, Zou et al. proposed intracellular transcription factor FOXP3 staining along with intracellular cytokine staining for IFNγ, IL-2, and tumor necrosis factor-α ,as anoptimized identification tool for characterization of nTregs based on the fact that activated conventional FOXP3+ T cells express these polyfunctional cytokines whereas the Treg cells do not thereby differentiating them from nTregs[26,27]. Nevertheless, the transient and fleeting FOXP3 expression of the conventional T cells brought problems in utilizing this transcription factor as a marker of Treg characterization. Approaches encompassing epigenetics emerged out to be an encouraging way to pass through this hurdle. Epigenetic modifications like DNA methylation plays a deciding role in a stable and sustained gene expression via chromatin remodeling. In this context it deserves special mentioning that a demethylated DNA sequence within the FOXP3 locus leads to a sustained FOXP3 expression[28].This specific methylation pattern differentiates Treg cells from other conventional T cells. Conventional T cells are specifically devoid of such specific demethylation pattern at the FOXP3 locus. Apart from this, recent researches have revealed the presence of CD127 as a potential marker to differentiate between Treg cell from the conventional T cells, the former being negative or dimly positive for CD127 which shows negative correlation with FOXP3 expression of Treg cells[29]. Therefore, majority of Treg cells can be characterized as CD4+CD25+CD127lowFOXP3+ T cells.
(b)Subsets of Regulatory T cells
Although Treg cells can be defined and classified as a separate and distinct subset of T cells endowed with suppressive attributes but current researched have depicted that this subset can be further categorized into various subsets depending on their phenotype function as well as developmental pathways. Current studies manifested that natural Treg cells arise from CD4-single positive thymocytes selection via MHC class-II dependent TCR interactions in the thymus. Simultaneously, under certain physiological conditions, adaptive or induced (iTreg) cells are generated outside the thymus[30,31]. Specific microenvironment enriched with cytokines like transforming growth factor-β (TGF-β) along with IL-2 create a favorable niche for both in-vitro as well as in-vivo generation of iTregs from naïve, resting T cells or other conventional T cells[32]. Till today, a vast array of phenotypically and functionally distinguished iTreg cells of both CD8 as well as CD4 lineages have been recounted for. The most notable subsets among them include CD4+CD25+ nTreg-like, CD8+CD25+ cells and IL-10 positive regulatory T cells (Tr1)[33].
(c)Mechanisms Encompassing the Suppressive Attributes
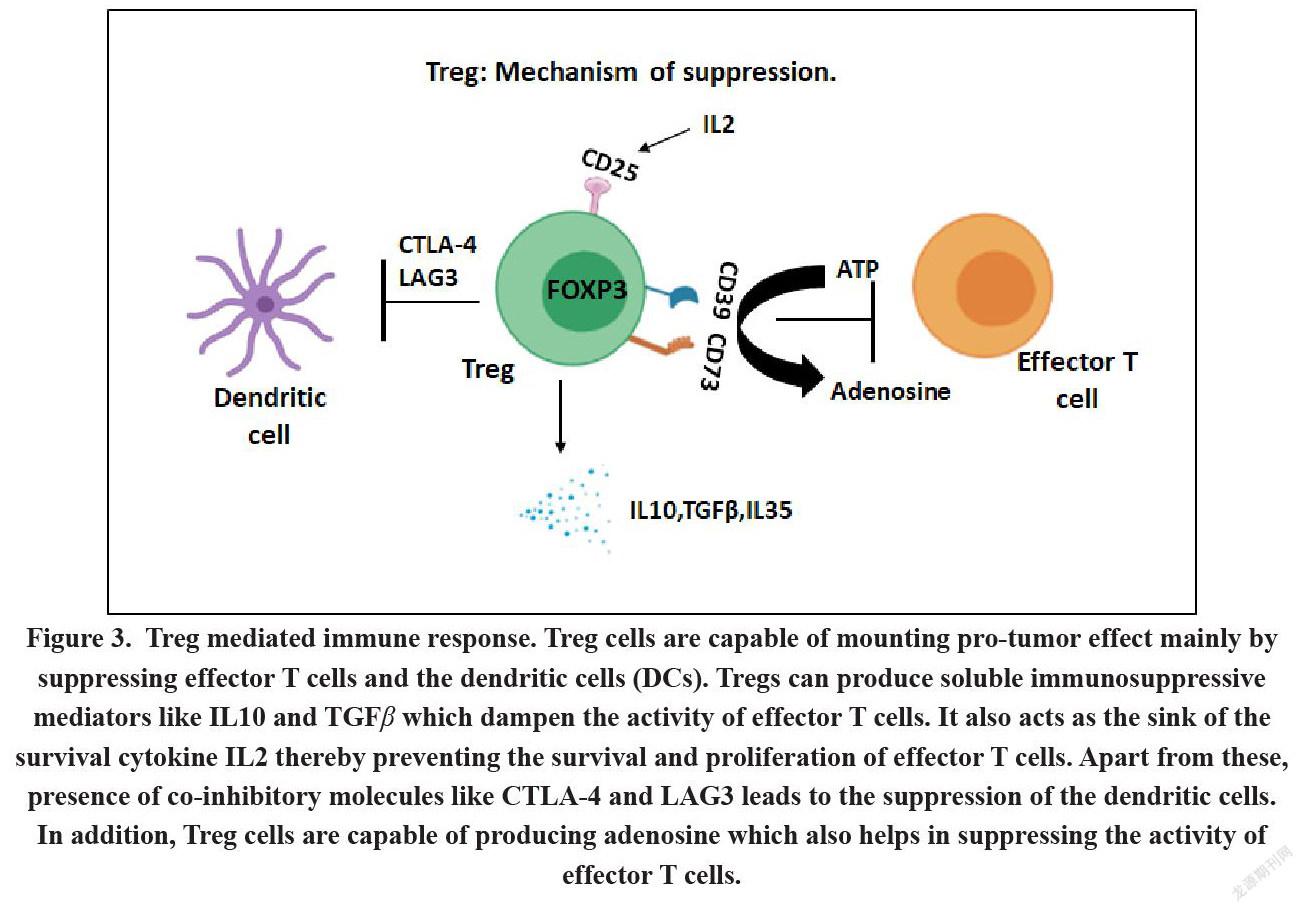
Treg cells play a distinguished part in cancer progression due to their ability to suppress the effective anti-tumor immune response. Treg cells mediated suppression of immune cells occurs either by cell-to-cell contact dependent mechanism or via contact independent mechanisms[34]. Proliferation of effector T cells as well as production of anti-tumor cytokines by effector T cells are inhibited via TCR activation of Tregs in a contact dependent manner thereby leading to IL2 inhibition[35]. Additionally, CTLA-4 and LAG-3 constitutively expressed on Tregs which lead to the cell to cell contact dependent suppressive mechanisms via their interaction with CD80 and CD86 expressed on the Antigen Presenting Cells (APCs)[36]. CTLA-4 on the other hand acts as a co-inhibitory molecule by directly competing with the co-stimulatory signal transduction pathway by acting as a ligand of higher affinity for both CD80 and CD86 than the stimulatory molecule CD28[37]. Likewise, CTLA-4 induces the expression of the enzyme IDO in the dendritic cells thereby directly suppressing the latter. IDO catalyzes the conversion of tryptophan to kynurenine thereby leading to tryptophan depletion and subsequent generation of immunosuppressive metabolites causing the impairment of the effector T cell function[38]. It has also been elucidated that CTLA-4 binding to CD80 and CD86 results in their dimmed expression on dendritic cells. This further resulted in the abrogation of T cell activation by dendritic cells leading to anomalous stimulation and generation of iTregs. LAG-3- another suppressive molecule expressed by Treg cells acts as a CD4 homologue which competitively binds to the MHC class II molecules expressed on APCs thereby leading to the suppression of effector T cells[39].In addition, Treg cells can also arbitrate the contact independent suppression mechanism via secretion of numerous soluble factors such as TGF-β or IL-10[39,40].Membrane-bound TGFβ present on Tregs cells play a deciding role in mediating Treg suppression of NK cells and T cells in a contact dependent manner. Recently researchers have identified another inhibitory cytokineIL35 from Treg cells of murine origin[41]. Additionally, expression of Galectin-1 belonging to a conserved family of β-galactoside-binding protein is observed on human Tregs. It binds to glycoproteins such as CD45, CD7 and CD43 as homodimer causing apoptosis, growth arrest and disruption of the secretion of pro-inflammatory cytokines from the activated T cells[42].
Abrogation of galectin-1 dampens the inhibitory efficacy of both murine as well as human Treg cells. Still it is not clearly known whether galectin-1 acts in a cell to cell contact dependent manner or as soluble factors in a contact independent manner[43]. COX-2 + iTregs also secrete suppressive prostaglandin E2 (PG)E2, and it has been observed that the presence of these cells in human colorectal cancer patients results in poor prognosis[44]. Depletion of these cells by the COX inhibitor indomethacin facilitate the T cell efficacy resulting in better prognosis and recurrence free survival of the patients. Apart from this, expression of ectonucleotidases like CD73 that by Treg cells has been recently reported. CD73 converts extracellular ATP to adenosine which in turn inhibits T cell function through the adenosine receptor 2A by causing cell apoptosis or by hampering the IFNγ secretion[45]. Another important feature of Treg cells that attributes to its immunosuppressive nature is its ability to act as the “sink” of IL-2. IL-2 is required for all T cells including Treg cells for proper maintenance of their function. While the conventional T cells are able to produce IL-2, Treg cells do not produce IL-2 themselves, instead they utilize their overtly expressed IL-2 R(CD25) to siphon off the IL2 produced by other T cells, thereby leading to the loss of function of T cells[32](Figure 3). Similar to this phenomenon of cytokine depletion, exhaustion of free thiol groups can also render an adverse effect on the activated T cells. Activated T cells require cysteine due to the lack of the transporters for its oxidized form-cystine. Dendritic cells generate a cysteine-endowed microenvironment via extra and intra-cellular redox reactions thus providing cysteine to T cells (Angelini et al., 2002)[46]. Treg cells impede this process by competitively consuming thiols including cysteine[47].Lastly, human Treg cells directly suppress the proliferation of responder T cells and initiate the DNA damage response by triggering glucose competition, ultimately resulting in senescence and functional changes in T cells[48].
4.2 Th17 cells: Paradoxical role in tumor immunity
(a)Characteristics, Regulation and Biological Functions
Th17 cells were recognized in the year 2005 by Harrington et al[49]. Gradually Th17 cells have set off huge interest in inspecting their regulation, development as well as their therapeutic manipulation in various pathophysiological conditions. Although IL-17A and IL17F acts as the signature cytokines of Th17 cells but numerous reports have shown that these cells can produce other cytokines like IL-22 or TNF-α[50]. Differentiation of naïve T cells results from a combination of different cytokines including IL-6, IL-1β and TGFβ (Langrish et al. 2005; Chung et al. 2006; Mangan et al. 2006; Veldhoen et al. 2006)[51].RORγt is the master transcriptional factor of Th17 cells which regulates the production of its signature cytokine IL-17A and IL-17F[52]. Th17 cells endowed with proinflammatory properties play an important role in several chronic inflammatory as well as autoimmune diseases[53]. However, later it was observed that Th17 cells under certain circumstances can also display potent regulatory and anti-inflammatory properties mainly via the production of immunosuppressive Treg specific cytokine IL-10[54]. These distinct type of Th17 cells are termed as regulatory Th17 cells. Further it was observed that this regulatory subset of Th17 cells co-express Treg cell specific transcription factor-FOXP3 as evidently observed in patients suffering from lung and colon cancer[55]. All these observations created a lot of perplexity about the nature as well as function of Th17 cells, especially their role in context of tumor immunopathology. It can be said, that preferential accumulation of Th17 cells in different types of tumor as compared to the healthy tissue might have different outcome with respect to tumor progression, depending on the tumor type[56]. In other words, Th17 cells can be associated with both the bad as well as good prognoses. However, it is worth asserting that the most confusing part arises from the fact that Th17 cell can exhibit high degree of plasticity. Th17 cells can transdifferentiate into Th1 cells as seen in numerous autoimmune diseases or into Treg cells as seen in several cancer types[57]. Apart from these, Th17 cells can also differentiate into TR1, Th2 or TFH cells thereby bestowing them with numerous opposing attributes and subsequently allowing them to generate qualitatively definite responses in context of varying microenvironment. In the next section, we will therefore try to discuss how the plasticity of Th17 cells and its developmental relationship with Treg cells influence the T cells present in Tumor microenvironment thereby affecting the tumor niche.
(b)Th17 cell: The FRIEND of Tumor
Unconventional immunosuppressive attributes shown by tumor specific Th17 cells might account for its pro-tumoral role in tumor microenvironment[56]. Th17 clones derived from human TILs characterized by Rorγt expression and IL-17 production when cultured in-vitro naturally gets converted into Treg cells upon TCR engagement expressing the Treg specific transcription factor FOXP3 and also acquiring the immunosuppressive properties[58]. It is important to note that this Th17 to Treg conversion is stable and not transient as the Treg cells derived from Th17 clones were rather obstinate to convert back to Th17 phenotype even if they were kept in presence of Th17 polarizing cytokine milieu[59].However, it is still a matter of debate whether Th17 cells can actually convert to Treg phenotype under in vivo condition in tumor microenvironment. However recent researches witnessed the existence of Th17/Treg hybrid IL17+FOXP3+ cells in numerous kinds of tumors. Though majority of the studies on this cell have shown that they originate from bona fide Treg cells but few contradictory reports have also shown these double positive cells to originate from the pool of Th17 cells present in tumor niche[54,60,61]. Interestingly these double positive cells have shown different outcomes in context of disease prognosis. For instance, these cells when extracted from the colorectal cancer biopsies, exhibit pro tumor role by promoting tumorigenicity in spheres that forms stem cells and also via the inhibition of tumor associated CD8+ effector T cells[62].Contrarily, the Th17 cells originating from Treg cells in a melanoma murine model, exhibit potent antitumoral effects[63]. IDO expressing plasmacytoid dendritic cells(pDCs) activated by CpG, prevents Treg conversion. It was observed that inhibition of IDO in pDCs resulted in IL6 production thereby promoting Treg plasticity towards Th17 cells[64].Therefore, the reports narrating Th17 plasticity in tumor microenvironment are very limited requiring further verification before determining their origin cell being either Th17 or Treg cell. In concurrence to potential cell plasticity, Th17 cells are capable of attributing immunosuppressive functions due to the presence of ectonucleotidases CD39 and CD73 on their surface. It has been well corroborated that production of adenosine successively by CD39 and CD73 imparts immunosuppressive properties to Treg cells mainly by inhibiting the proliferation of T cells and interference with the cytokine production ability of the cells[65]. In-vitro generation of Th17 cells in presence of cytokines, TGF- β+IL-6 results in the expression of the ectonucleotidases CD39 and CD73 on their surface, while classical Th17 cells devoid of the aforementioned immunosuppressive molecules are generated in presence of the cytokines IL-6, IL-23, and IL-1β [66]. Concomittant expression of the ectonucleotidases CD39 and CD73 creates a purinergic hallow surrounding the immune cells thereby conferring the immunosuppressive attributes to Th17 cells. These in-vitro generated ectonucleotodases-expressing Th17 cells, when transferred in to murine models, promptly resulted in tumor development. Surprisingly, those cells were not showing the presence of Foxp3 thereby not representingTh17 to Treg transition phenomenon and thus can be termed as bona fide Th17 cells. Taken together it can be concluded that Th17 cells, depending on the cytokine stimuli that it encounters, can support tumor development mainly by accelerating angiogenesis and/or via the inhibition of anti-tumor immune responses.
(c)Th17 cells: Foe of tumor
In addition to the protumor activity mediated by Th17 cells, several clinical correlation studies and mounting evidence in cancer models have suggests that Th17 cells may also have potent antitumoral immune effects. Murine tumor models have shown that tumor growth was increased in both IL-17−/− (B16 melanoma and MC38 colon cancer cell lines) and Rorγt ?/− mice (B16 melanoma cell line)[67]. Muranski et al showed Th17 cells have the potential to reduce advanced and established B16 melanoma in murine models[56]. In IL-17−/− murine models, profound tumor growth and lung metastases were associated with decreased IFN-γ+ NK cells and IFN-γ+ T cells in tumor draining lymph nodes and, in the tumor, itself implying that Th17 cell associated therapeutic effect was essentially dependent on IFN-γ production[68]. Furthermore, Hinrichs et al reported that adoptive transfer of in-vitro polarized Th17 cells resulted in regression of established melanoma or reduction in number of tumor foci in melanoma mice model[69]. Apart from this, plasticity of Th17 cells towards Th1 phenotype also contributes to its anti-tumor efficacy. It was observed that transferred IL-17-producing CD8+ T cells gets converted into IFN-γ-producing effector T cells thereby mediating antitumor effects[65]. In addition to direct eradication of tumor cells, Th17 cells adopt several mechanisms to have an indirect anti-tumor effect. It was shown that Th17 cells can contribute to anti-tumor effect by inducing the Th1 specific chemokines like CXCL9 and CXCL10 promoting the recruitment of effector T cells in tumor site[70]. In addition, tumor specific Th17 cells also induce CCL20 production that facilitates the recruitment of DCs and their consequent migration to the draining lymph nodes of tumor leading to activation of CD8+ T cells. It was also reported that Th17 cells accumulation in human malignant pleural effusions envisaged improved survival of patients. Additionally, in lung cancer patients, MAGE-A3 tumor antigen-specific human Th17 cells converted into IFN-γ secreting cells during differentiation into effector T cells thereby mediating potent antitumor effects in vivo[71].These data, thus, suggest the prospective protective anti-tumor function of human Th17 cells in context of tumor immunity.
4. Immunological Fate of Effector T Cell Favorable for Tumor Progression
Incessant exposure to tumor specific antigen is one important factor that renders the T cell dysfunction in tumor microenvironment. Dysfunction T cells are mainly characterized by their inability to show profound proliferation along with their reduced effector function and overexpression of numerous inhibitory molecules. The presence of an enriched inhibitory network in tumor microenvironment, as described previously, leads to distinct dysfunction states of the tumor specific T-cells[11,21]. Therapeutic interventions causing the effective reactivation of Tumor specific T-cells resulted in a better prognosis of cancer patients. The upcoming sections focus on the hallmarks of T cell dysfunction in cancer. Also, we discuss the role played by the immune system itself for T cell dysfunction and the relationship between T cell dysfunction and cancer immunotherapy (Figure 4).
(a) T Cell Anergy
T cell anergy refers to an induced condition of functional inactivity of T cells following antigen exposure[11,72]. However, the cells remain alive for a long period of time in a hypo responsive status as described by reduced co-stimulation and/or expression of higher extent of co-inhibitory receptors. A functional interpretation proposes that T cell anergy results from the process of immune regulation and it is incited on the peripheral resting/naïve T cells for augmenting tolerogenicity against self-antigens and protection of the host system from autoimmune disorders[73]. Human tumors endowed with a powerful immunosuppressive network provides an ideal microenvironment for T cells to be under the exposure of co-inhibitory molecules leading to T cell anergy in the tumor[21,74]. Notably in tumor microenvironment, immune suppression of T cell is prompted by interaction with potent co-inhibitory receptors like PD-L2, PD- L1, ICOS-Land B7-H4 [75]. Alongside, negligible CD80 and CD86 co-activatory receptors detection from the cells in tumor microenvironment or tumor infiltrating Antigen presenting cells (APC) further facilitates the process[21]. This active disproportionateness between lowered co-stimulatory but increased co-inhibitory receptors leads T cells in favor of immune unresponsiveness.
(b) T Cell Exhaustion
T cell exhaustion can be described as a progressive loss of the effector function of T cells mainly characterized by lowered cytokine expression and also by their inability to undergo reactivation upon receiving stimulatory response[76]. This mainly describes those effector T cells which exhibits strong and repetitive activation in case of certain persistent inflammatory conditions like cancer, chronic infection. Recurrent T cell stimulation leads to their procurement and expression of numerous inhibitory molecules on their surface as observed in case of several chronic infection as well as cancer. Ligation of PD-1 with its ligand leads tothe association of SH2- domain to the immunoreceptor tyrosine-based switch motif (ITSM) present within the PD-1’s cytoplasmic tail[77]. This in turn leads to the inhibition of the TCR signaling pathways and subsequently inhibits T cell stimulation.
(c)Senescent T cell
Cellular senescence can be defined as a naturally occurring physiological process due to cell proliferation[11]. Lack of CD28 expression, telomere shortening and also the inability to enter cell cycle appears to be the chief attributes that characterize T cell senescence[78]. Apart from this, senescent T cells also acquire certain immunosuppressive as well as dysfunctional killing abilities[21]. In addition, phenotypic analysis showed a high level of expression of CD57, Tim-3 as well as killer cell lectin-like receptor subfamily G, member 1 (KLRG-1) on senescent T cells[79]. Interaction of these cells with galectin-9+ tumor associated myeloid Antigen presenting cell were also observed[80]. All these findings were further observed in lymphoma and melanoma patients suggesting that Tim-3+ cd28lo T cells present in cancer may contain senescent T cells that ultimately succumb to cell cycle arrest.
(d)Polyfunctional T cell
Immune cells are normally classified and defined on the basis of the expression of surface chemokine receptors as well as specific transcription factor(s). In addition, immune cells can also be classified on the basis of the cytokine(s) that they secrete. For instance, Th1 can be classified by the secretion of signature cytokine IFNγ, Th2 via secretion of IL-4, Treg via secretion of IL-10 and Th17 via secretion of IL-17. However, lymphocytes, under certain condition, produce multiple cytokines and are referred to as the “polyfunctional T cells” which can mount better immune response than their monofunctional counterparts (producing one type of cytokine). Most of the researchers advocate for the effective anti-tumor immune response of polyfunctional T cells (discussed elaborately in the later section of the article) but few reports suggest otherwise. As reported by Ding et. al, adoptive transfer of CD4+T cells in combination with chemotherapy, results in initial regression of tumor. However, this combinatorial method is not durable and ultimately results in relapse. Notably, the relapse is associated with the acquisition of tolerized phenotype in CD4+ T cells mainly via the overexpression of PD-1(Programmed death-1). Effective blocking of PD-1-PDL-1 pathway can successfully alleviate this pro-tumor role of the polyfunctional T cells.
(e) T cell apoptosis
Tumor cells can effectively reduce the number of anti-tumor immune cells in its microenvironment via the expression of FasL, which induces apoptotic death of cells expressing the Fas receptor (Fas) This phenomeno has been demonstrated on the tumor vasculature in ovarian cancer and other tumor types which results in the reduction of T cell infiltration. In Pancreatic ductal adenocarcinoma, FasL is additionally expressed by tumor cells, thereby implying that Fas-dependent induction of effector T cell death can be utilized by multiple cell types in the TME.
6.Targeting T Cells for Mounting an Anti-tumor Response
In this section we tried to illustrate the ways of successful harnessing of immunotherapeutic drivers for mounting an effective anti-tumor response. Two major therapeutic drivers broadly encompass immune checkpoint blockade (ICB) therapy using antibodies blocking inhibitory receptors of the immune system across tumors and adoptive cell therapy (ACT) using T cells engineered to express chimaeric antigen receptors (CAR T cells) targeting blood malignancies.
6.1 T Cell Stemness
Stem cells refer to a distinctive subset of cells that possess both multi-lineage differential potential as well as self-renewal capacities. T cell stemness arises out to be a latest conception and in this arena, role of memory T cells deserve special mention. Memory T cells possess increases self-renewal capacities and are also able to generate more differentiated forms of memory T cells[81]. These “T memory stem cells” are rarely observed in the peripheral blood of healthy volunteers. Human and murine tumors suggest that Th17 cells present there may reflect stemness characteristics as discussed in the next portion. Nevertheless, it has been observed that central memory T cells from murine origin have escalated stemness properties as these got arrested at a pre-differentiated state and have a soaring potential for the production of T effector cells following exposure to secondary antigen challenge[82]. T memory cells possessing high stem-cell like properties lead to the ceaseless development of T effector cells throughout the entire human lifetime thereby ensuring the continuous replenishment of differentiated T cell pool despite the finite lifespan of the individual differentiated T cells present in the periphery[83]. Recent researches in murine models show the presence of some CD44 low CD62L high memory CD8+T cells[84]. These cells possess stem cell signature gene namely stem cell antigen-1 (Sca-1). Additionally, these cells possess increased self-renewal properties and the capability to produce effector as well as central memory T cells. It has been observed that memory T cells are generated in-vivo via the co-engagement of CD3 and CD28 in the context of IL15 and IL7[85]. Though IL7 and IL15 plays crucial role in imparting stem-like property to T cells but not all IL7 and Il15 activated T cells acquire stemness. Additional factors are required for the impartment of stemlike properties in T cells. In this context the presence of IL21 deserves mention. It has been elucidated that IL21 restrains T cell differentiation by sustaining the expression of WNT-β-catenin transcription factors TCF7 and LEF1 thereby facilitating generation of T cells with stem cell like properties [90]. Thus, the transcription factors TCF7 and LEF1 are crucial for the development as well as maintenance of T-cell stemness and the TSCM (Stem memory T cell) phenotype. It has been further elucidated that blockade of T cell differentiation results in Wnt signaling expression and subsequent generation of T cells with stem cell like properties[86]. In summary, it can be said that a better understanding of the stemness property of T memory stem cell population and their ability to promote their production in order to maintain the pool of self-renewing, long lived and antigen experienced memory T cells might provide a beneficial therapeutic approach for cancer patients.
6.2 Th17 Cells
As mentioned in the previous section, numerous recent researches reported the existence of a group of Th17 cells possessing increased stem cell markers in human tumors. Mouse and human Th17 cells are shown to have increases survival potential, persistence and also possess the ability to repopulate sublethally irradiated murine models[87]. Additionally, these cells reflected potent anti-tumor responses in comparison to the central and effector memory T cells. While studying the underlying signaling pathway, it has been observed that HIF1α/ NOTCH/Bcl-2 plays an important role in mediating their stem-cell specific characteristics[88]. It can also be concurred that stem cell lineage may involve better immune responses. Supporting this notion, Th17 stem cell-like T cells possess the capability to differentiate into distinct Th subsets mainly moving towards either Th1 or Treg like cells[89]. Apart from this, stem-like Th17 cells possess greater survival capabilities as well as self-renewal properties. Human Th17 cells residing in tumor can be described as long-lived cells possessing increased self-renewal capabilities and are able to differentiate into highly effective effector T cells. Thus, manipulation of the stem-like properties of Th17 cells might be of therapeutic interest in treating malignancies.
6.3 Polyfunctional T Cells
Polyfunctional T cells in context of tumor immunology refers to a group of tumor reactive effector T cells capable of mounting greater immune responses[90]. These cells are characterized by the transient expression of multiple cytokines which act as the hallmarks of effective immune response. Polyfunctionality of T cells can be defined on the basis of their ability to produce cytokine specific for either CD4+ or CD8+ T cells mainly IL-2, TNF-α, IFN-γ or Granzyme-B, TNF-α and IFN-γ respectively[91]. Role of these T cells in developing potent immune response is well documented in context of several viral infections like vaccinia virus and HIV[92]. Ability of the polyfunctional T cells for the co-expression of multiple cytokines enable them to concomitantly stimulate numerous pathways in the immune response. For instance, polyfunctional CD4+ Tcell expresses IL-2 which helps in the T cell survival during an ongoing immune response; simultaneous expression of IFN-γ promotes inflammation in the surrounding while TNF-α inhibits viral replication by successfully activating the adjacent epithelium tissue[93]. On a similar account, polyfunctional CD8+ T cells expressing IFN-γ, Gra-B, and TNF-α mediates the directed cytolysis of the infected cells[94].
Such responses have shown a better and efficient pathogen clearance under different pathophysiological conditions. However, studies related to polyfunctional T cells in context of tumor are scarce. Recent study has shown that Th17 cells present in human ovarian cancer synergistically produce both IL-17 as well as IFN-γ. Expression of these cytokines in turn stimulated the production ofchemokine factors like CXC10 and CXCL9 from the adjacent tumor cells. Finally, this chemokine gradient leads to the recruitment of effector T cells in tumor niche thereby mounting an effective anti-tumor response. Additionally, in murine model, chemotherapeutic treatment against B-cell lymphoma resulted in induction of polyfunctionality of T cells[95]. T cell infiltration in tumor microenvironment often results in the suppression of the T cell responses. Thus, inhibition of these immunosuppressive molecules leads to an enhanced response of the polyfunctional T cells in tumor microenvironment. On a specific term, CTLA-4 inhibition in melanoma patients resulted in the co-expression of the cytokines IFN-γ, MIP-1β, and TNF-α which in turn mount an effective anti-tumor response[96]. However, the exact phenotypical definition of polyfunctional T cell subset is still an active field of research. Till date it has been elucidated that this attribute is a transient phase of activated T cells achieved via TCR mediated signaling cascade. Further it has been observed that single cytokine expressing monofunctional T cells might give rise to these polyfunctional subset of T cells. It still remains a question to ascertain whether these polyfunctional T cells are merely the product of robust activation or there lies some genetic signatures that define this T cells in a pre-decided manner.
6.4 Regulatory T Cells
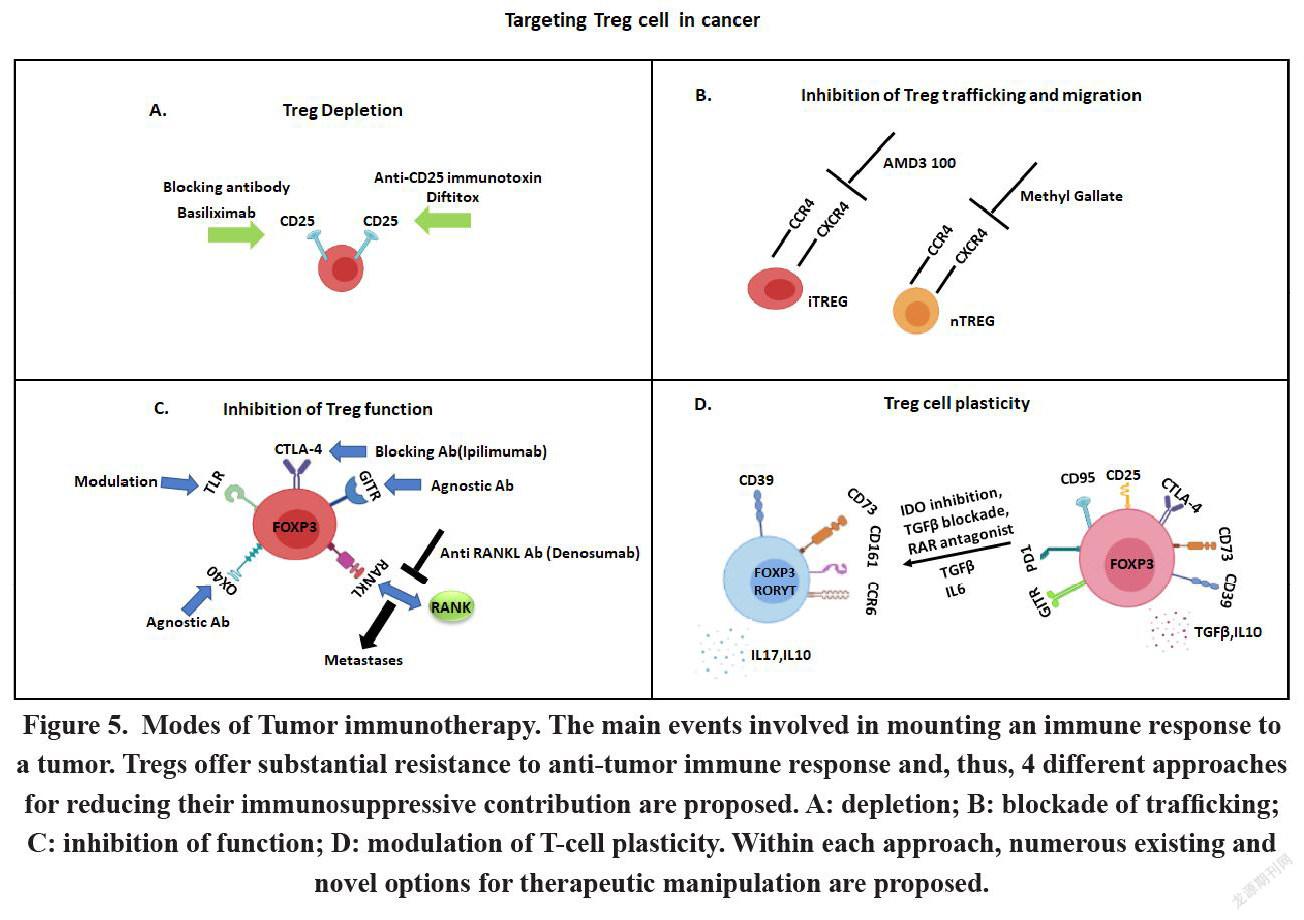
Regulatory T cells (Tregs) irrespective of their origin and site of migration, hamper and dampen tumor surveillance and in numerous circumstances turns out to be one of the major contributing aspects for tumor progression. Robert North and colleagues, back in 1980, have shown that tumor cells get sensitive to immune reaction by depleting Treg cells[97]. However, modulation in Treg cells might have some serious side effects particularly in context of autoimmune diseases. Additionally, it is of utmost importance to find out ways for the depletion of Treg cells exclusively from the site of malignancies as its global deletion might have some deleterious effect on the physiological condition. Numerous approaches involving the depletion of Tregs or functional inactivation of Tregs are currently under development or undergoing evaluation at clinical stage. Tregs present within the TILs, express numerous cell surface markers, including CD25, CTLA-4, GITR, OX40, and mAbs against these cell surface markers can potentially cause the inhibition of Treg cell function [98]. Recent studies have revealed if CD25+CD4+Tregs are removed and inactivated by anti-CD25 mAb or toxin-conjugated anti-IL-2 (Denileukin diftitox) respectively, Teff cells get activated, that leads to inhibition of tumor growth in rodents[98]. Anti-CD25 mAb for Treg depletion has shown potent outcome and is therefore undergoing clinical trials. In one research study, it has been found breast cancer patients those who have received vaccination against tumor associated antigens along with administration of an anti CD25 mab called daclizumab- shows a median progression free survival of 4.8 months in 6 out of 10 patients, due to hyperactivation of T cells. Another research work in patients with melanoma contradicts the previous study by showing that administration of daclizumab to deplete Treg cells is not effective in mounting antitumor immunity. They observed that not only Treg cells get depleted but Teff cells also get depleted during this treatment and the reason behind this outcome is when Teff cells are activated they also start to express CD25 cell surface marker. So targeting CD25 to deplete Treg cell is not a good approach to augment antitumor immunity. [98]
Monoclonal antibody Ipilimumab and tremelimumab (CTLA-4 blocker) against the immune-checkpoint molecule-CTLA-4, as expressed by activated CD4+ and CD8+ Teff cells as well as FOXP3+CD4+ Tregs, has been shown to facilitate effective tumor suppressing attribute[99]. The mechanism of action of this mab is previously thought to be dependent on the interference between CTLA4 and CD80/ CD86 in Teff cells and APC, which in turn helps in the bypassing of natural inhibition process of T cells after getting activated. However, recent preclinical studies indicated a different mode of action performed by this mab to promote anti-tumor immunity via the depletion of CTLA4 expressing Tregs by antibody dependent cell mediated cytotoxicity (ADCC) thereby resulting in higher ratio of Teff to Treg in the tumor microenvironment. Disruption of the Fc region of the mab completely diminish its antitumor immunity as the Fc region of the Ab facilitates the process of ADCC. The major problem of this immunotherapy is diverse side effects during and after the treatment, mainly development of colitis characterized by high rate of colonic ulcerations.
Elevated expression of several Co-stimulatory receptors like GITR, OX40, and ICOS, by Treg cells emerges out to be potent targets for the successful depletion of Treg cells resulting in the depletion of their function[100]. Differential expression of GITR in resting CD4 and CD8 T cells and in Treg cells can be a good candidate for immunotherapy. In resting CD4 and CD8 cells expression of GITR is low compare to Treg cell which express high level of GITR. When GITR signalling pathway is activated in Treg cells by using an agonistic anti-GITR mAb, suppression activity of Treg cells hamper thereby promoting the induction of Treg-resistant Teff cells. The GITR agonists are now being investigated as potential candidate for treatment of numerous solid cancers at their advanced stage.
Similarly, OX40-agonists, engaging the OX40 molecule that is being constitutively expressed by a subset of Tregs, stimulate anti-tumor responses of Teff cells, but the effect of this therapy on Tregs in cancer is still partially explored thereby leaving the requirement for more rigorous research. Immunotherapy involving OX40 agonist alone or in association with other mab are being looked into in patients with solid cancer or melanoma. Activated Treg cells in tumor-infiltrating lymphocyte (TIL) of patients with gastric cancer, shows elevated expression of ICOS thereby controlling the function and homeostasis of Treg cell. ICOS agonists supposedly have a dual-mode of function i.e. activation of Ag-specific CD4+ Teff cells and concomitant depletion of Tregs[98].
A new way of immunotherapy is emerging which involves targeting intracellular signalling in Tregs for mounting anti-tumor response. One of the promising Treg directed therapy is to target PI3K signalling pathway, that helps in Treg maintenance and function. It has been found inhibitors of PI3K reduces the suppressive property of Treg in mouse models. Inactivation of PI3Kδ in Tregs leads to the increased activity of CD8+ T cells thereby leading to tumor. Functional disruption of the PI3K-phosphatase and tensin homolog (PTEN)-mTOR pathway in Tregs leads to the impairment of the mitochondrial activity, thereby leading to the disruption in FOXP3 expression of Treg cells, simultaneously increasing the activity of Teff cell. Combinatorial treatment involving administration of pembrolizumab and PI3Kδ inhibitors is recently emerging out to be a potent candidate of phase I trial in patients suffering from advanced stage of solid tumors. Also, treatment of chronic myeloid leukemia affected patients with tyrosine kinase inhibitors, including imatinib and dasatinib, showed reduction in Treg survival and function through off-target effects. Another mechanism of Treg mediated immmunesuppression is via the production of suppressive molecule adenosine by the help of CD39 and CD73 which are expressed by this cell. Therefore, CD39 and CD73 could be a promising target for therapeutic usage. All the cancer immunotherapies that target Treg associated molecules for augmentation of anti tumor immunity have some adverse side effects due to off target, specifically targeting the whole compartment of Treg in the system (Figure 5A-D). New technological advancements, like using nanoparticles as carrier for Treg-targeting molecules at particular compartment like tumor microenvironment can increase the efficacy of the therapy without any major side effect ultimately better prognosis of the patients[101].
6.5 CAR T Cells: Immune Cells engineered to Fight against Cancer
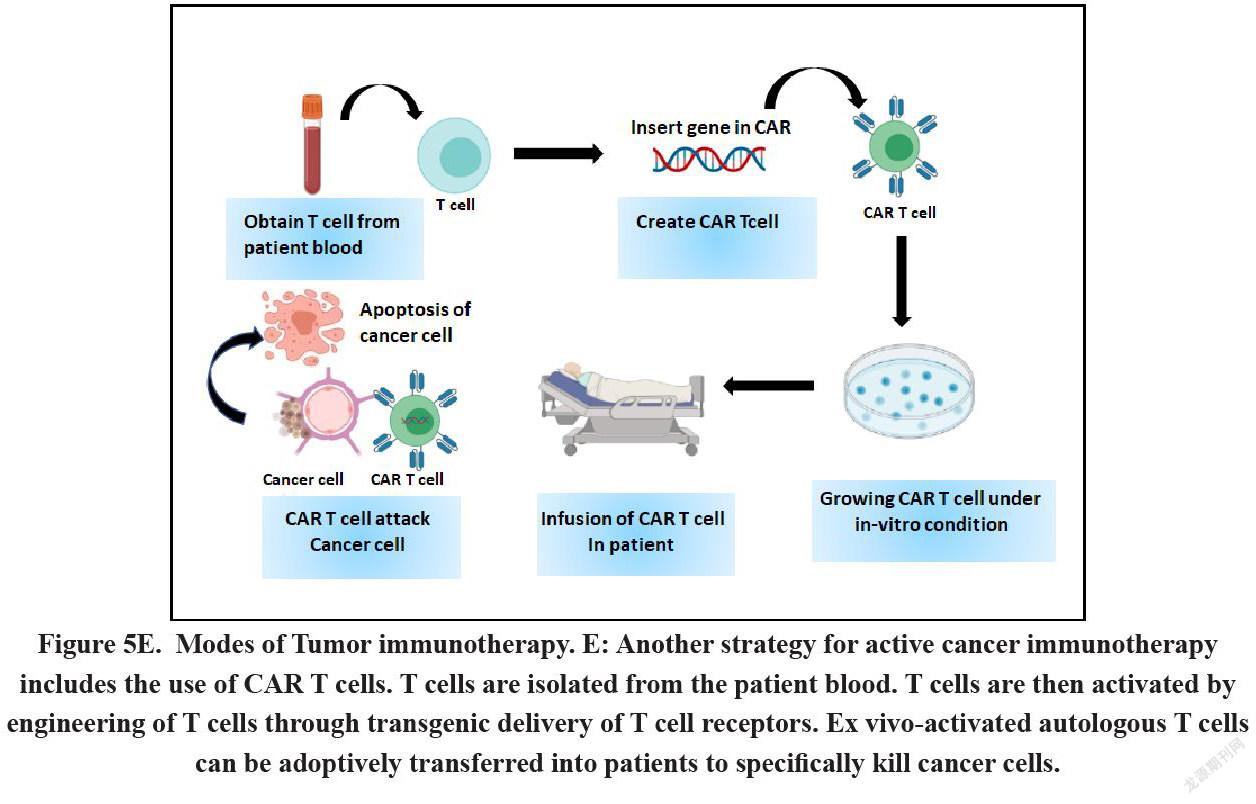
Chimeric Antigen Receptor (CAR) T-cell therapy refers to the alteration of autologous T-cells isolated from patients in genetic level so that the cells can show the expression of a CAR specific for a tumor associated antigen[102]. The genetically modified cells are then expanded under ex vivo condition following their re-introduction into the patient. CARs can be defined as fusion proteins comprising of a selected single-chain fragment variable from a specific monoclonal antibody and one or more T-cell receptor intracellular signaling domains. In general, viral-based gene transfer methods or certain non-viral methods like CRISPR/Cas9 technology, DNA-based transposons or direct transfer of in-vitro transcribed mRNA by electroporation are the normally followed methods to induce the genetic modification in the cells. Clinical trials have given a far-fetched satisfactory result in the advanced stage patients suffering from hematological cancers like Acute Lymphocytic Leukemias (ALL). However, the efficacy of this therapy is far less promising in case of solid tumors due to numerous therapeutic barriers like the persistence, expansion fate and trafficking of CAR-T cells within tumor (Figure 5E).
6.6 Targeting Myeloid Derived T Cells to Elicit Anti-tumor Response
Myeloid-derived suppressor cells (MDSCs) comprising of different cell types including granulocytes, macrophages and dendritic cells, possess the ability to abrogate the immune response of numerous anti-tumorigenic immune cells through direct or indirect mechanisms. Both monocytic and granulocytic MDSCs are recruited in the tumor microenvironment by tumor-shed soluble factors. The monocytic MDSCs utilizes NO and arginase whereas the latter utilizes ROS (Reactive oxygen species) to exert their immunosuppressive functions. Monocytic MDSCs play the key role in suppressing T cells in tumor microenvironment via upregulation of FOXP3 expression in T cells and also by directly disrupting the antigen specific CD8+ T cell responses via nitration of tyrosines in a T-cell receptor TCR-CD8 complex, thereby affecting its interaction with Pmhc. Recent reports suggest that MDSC down-regulate L-selectin expression on naive T cells, thereby preventing their homing to the activation site. Thus, successful targeting of MDSCs can be a potent way to elicit successful anti-tumor immune response.
7.Concluding Remarks
Recent researches are putting a lot of focus on elucidating the signaling pathways related to T cell dysfunctions and also on the cellular and molecular players that enforces such conditions on T cells. Recent works have recorded intense immune suppressive disparity in the tumor microenvironment like the ample existence of co-inhibitory molecules, the absence of co-activatory molecules and also the increased presence of immunosuppressive cells like Tregs or MDSC. Numerous clinical trials in the field of cancer immunology aim in shifting the microenvironment endowed with high suppressive network towards the condition described by the presence of lowered co -inhibitory molecules and lesser immunosuppressive cells through antibody blocking of specific co-inhibitory molecules or by attacking the immunosuppressive cells. There is a persuasive amount of affirmation in support of the concomitant existence of T cell anergy, senescence, exhaustion as well as stemness and polyfunctionality in context of tumor microenvironment. With further scientific advancement, new ways will be explored for identifying specified functional characteristics of T cells. Present literature manifests Tim-3 + and KLR1+ represent senescent T cells, PD-1+ T cells represent functionally exhausted effector T cells while Sca-1+ T cells may be stem-like T cells of murine origin. Nevertheless, as we have investigated, these markers do not circumscribe their specific prototype. Probably, these T cell paradigms are generated in a functional context depending on their specific microenvironment and that is determined by specific functional and genetic pattern and not by mere surface phenotypes. Th17 cells act as a useful example in this context. These cells have increased self-renewal properties but simultaneously express surface markers specific for terminal differentiation. A second example described that T cell dysfunction does not strictly follow phenotypic characteristics. It was observed that PD-1+ cells might show the expression of molecules like LAG-3 and Tim-3. Probably B7-HI/PD-1 and Tim-3/Galectin-9 mediated signaling pathways may act concurrently and/or additively to promote the dysfunctionality of T cells. Therefore, blockade of these molecules might play an important role in improving T cell mediated immune response in tumor microenvironment. Preclinical and clinical studies propose that dysfunctionality of T cells may be a functionally reversible phenomenon. Anti -tumor immunity mainly depends on the effective stimulation of responses mediated by T cells. Th17 cells representing a highly immunogenic subset of T cells possess increased polyfunctional capabilities as well as stem cell characteristics. On the basis of this knowledge development of certain strategies that enables the promotion of Th17 cell acquisition with the concomitant depletion of Treg cells from tumor microenvironment should result in better prognosis of the patients. Additionally, certain approaches that aim to the shifting of the immune response from Treg cells to Th17 cells should be looked into. In summary, tumors are able to seize mechanisms encompassing the T cell tolerance from the periphery that includes regulatory T cells, T cell anergy, senescence as well as T cell exhaustion, which in turn enable them to escape immune surveillance and survive. A better and deeper comprehension of the occurrence of these events will facilitate the establishment of successful therapeutic approaches thereby targeting human tumors. One such action plan may be promotion of T cell stemness and polyfunctionality along with the concurrent relief of the dysfunctionality of effector T cells.
References
[1] Roma-Rodrigues, C., Mendes, R., Baptista, P. V. & Fernandes, A. R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci, 2019, 20(4).
[2] Cooper, G. M. The Development and Causes of Cancer. Cell Mol. Approach 2nd Ed, 2000.
[3] Dvorak, H. F. Tumors: Wounds that do not heal--Redux. Cancer Immunol. Res, 2015, 3. 1-11.
[4] Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene,2008, 27, 5904–5912.
[5] Whiteside, T. L. Immune responses to malignancies. J. Allergy Clin. Immunol. 2010, 125, S272–S283.
[6] Dunn, G. P., Old, L. J. & Schreiber, R. D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360.
[7] Roles of the immune system in cancer: from tumor initiation to metastatic progression. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6169832/.
[8] Seung, L. P., Seung, S. K. & Schreiber, H. Antigenic cancer cells that escape immune destruction are stimulated by host cells. Cancer Res. 1995, 55, 5094–5100.
[9] Fouad, Y. A. & Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017,7, 1016–1036.
[10] Greten, T. F., Mauda-Havakuk, M., Heinrich, B., Korangy, F. & Wood, B. J. Combined locoregional-immunotherapy for liver cancer. J. Hepatol. 2019,70, 999–1007 .
[11] Thommen, D. S. & Schumacher, T. N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562.
[12] Liechtenstein, T., Dufait, I., Lanna, A., Breckpot, K. & Escors, D. MODULATING CO-STIMULATION DURING ANTIGEN PRESENTATION TO ENHANCE CANCER IMMUNOTHERAPY. Immunol. Endocr. Metab. Agents Med. Chem.2012, 12, 224–235.
[13] Okoye, I. S., Houghton, M., Tyrrell, L., Barakat, K. & Elahi, S. Coinhibitory Receptor Expression and Immune Checkpoint Blockade: Maintaining a Balance in CD8+ T Cell Responses to Chronic Viral Infections and Cancer. Front. Immunol. 2017,8.
[14] Teague, R. M. & Kline, J. Immune evasion in acute myeloid leukemia: current concepts and future directions. J. Immunother. Cancer 2013,1, 13.
[15] Bercovici, N. & Trautmann, A. Revisiting the role of T cells in tumor regression. Oncoimmunology , 2012,1, 346–350.
[16] Zamarron, B. F. & Chen, W. Dual Roles of Immune Cells and Their Factors in Cancer Development and Progression. Int. J. Biol. Sci. 2011,7, 651–658.
[17] Muz, B., de la Puente, P., Azab, F., & Azab, A. K.. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckland, N.Z.), 2015,3, 83–92. https://doi.org/10.2147/HP.S93413
[18] Meng, S., Li, L., Zhou, M., Jiang, W., Niu, H., & Yang, K.. Distribution and prognostic value of tumor infiltrating T cells in breast cancer. Molecular medicine reports, 2018, 18(5), 4247–4258. https://doi.org/10.3892/mmr.2018.9460
[19] Direct activation of antigen-presenting cells is required for CD8+ T-cell priming and tumor vaccination. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3198339/.
[20] Kaiko, G. E., Horvat, J. C., Beagley, K. W. & Hansbro, P. M. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology 2008, 123, 326–338.
[21] Xia, A., Zhang, Y., Xu, J., Yin, T. & Lu, X.-J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019,10.
[22] Murray, T. et al. Very Late Antigen-1 Marks Functional Tumor-Resident CD8 T Cells and Correlates with Survival of Melanoma Patients. Front. Immunol. 2016,7.
[23] Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4009604/.
[24] Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases - PubMed. https://pubmed.ncbi.nlm.nih.gov/7636184/.
[25] Baecher-Allan, C., Viglietta, V. & Hafler, D. A. Human CD4+CD25+ regulatory T cells. Semin. Immunol. 2004,16, 89–98.
[26] Devaud, C., Darcy, P. K. & Kershaw, M. H. Foxp3 expression in T regulatory cells and other cell lineages. Cancer Immunol. Immunother. CII 2014, 63, 869–876.
[27] Plitas, G. & Rudensky, A. Y. Regulatory T Cells: Differentiation and Function. Cancer Immunol. Res. 2016, 4, 721–725.
[28] Nair, V. S., Song, M. H., Ko, M. & Oh, K. I. DNA Demethylation of the Foxp3 Enhancer Is Maintained through Modulation of Ten-Eleven-Translocation and DNA Methyltransferases. Mol. Cells 2016, 39, 888–897.
[29] Liu, W., Putnam, A. L., Xu-Yu, Z., Szot, G. L., Lee, M. R., Zhu, S., Gottlieb, P. A., Kapranov, P., Gingeras, T. R., Fazekas de St Groth, B., Clayberger, C., Soper, D. M., Ziegler, S. F., & Bluestone, J. A. . CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. The Journal of experimental medicine, 2006,203(7), 1701–1711. https://doi.org/10.1084/jem.20060772
[30] Bilate, A. M. & Lafaille, J. J. Induced CD4+Foxp3+ Regulatory T Cells in Immune Tolerance. Annu. Rev. Immunol. 2012, 30, 733–758.
[31] Povoleri, G. A., Scottà, C., Nova-Lamperti, E. A., John, S., Lombardi, G., & Afzali, B.. Thymic versus induced regulatory T cells - who regulates the regulators?. Frontiers in immunology, 4, 169. https://doi.org/10.3389/fimmu.2013.00169
[32] Cheng, G., Yu, A. & Malek, T. R. T cell tolerance and the multi-functional role of IL-2R signaling in T regulatory cells. Immunol. Rev. 2011,241, 63–76.
[33] Gol-Ara, M., Jadidi-Niaragh, F., Sadria, R., Azizi, G. & Mirshafiey, A. The Role of Different Subsets of Regulatory T Cells in Immunopathogenesis of Rheumatoid Arthritis. Arthritis 2012.
[34] Thornton, A. M., & Shevach, E. M. (1998). CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. The Journal of experimental medicine, 188(2), 287–296. https://doi.org/10.1084/jem.188.2.287
[35] Paluskievicz, C. M. et al. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10.
[36] Sojka, D. K., Huang, Y.-H. & Fowell, D. J. Mechanisms of regulatory T-cell suppression – a diverse arsenal for a moving target. Immunology 2008, 124, 13–22.
[37] Sansom, D. M. CD28, CTLA-4 and their ligands: who does what and to whom? Immunology 2000, 101, 169–177.
[38] Zhai, L., Bell, A., Ladomersky, E., Lauing, K. L., Bollu, L., Sosman, J. A., Zhang, B., Wu, J. D., Miller, S. D., Meeks, J. J., Lukas, R. V., Wyatt, E., Doglio, L., Schiltz, G. E., McCusker, R. H., & Wainwright, D. A.. Immunosuppressive IDO in Cancer: Mechanisms of Action, Animal Models, and Targeting Strategies. Frontiers in immunology, 11, 1185. https://doi.org/10.3389/fimmu.2020.01185
[39] Corthay A.. How do regulatory T cells work?. Scandinavian journal of immunology, 2009, 70(4), 326–336. https://doi.org/10.1111/j.1365-3083.2009.02308.x
[40] Schmidt, A., Oberle, N., & Krammer, P. H. (2012). Molecular mechanisms of treg-mediated T cell suppression. Frontiers in immunology, 3, 51. https://doi.org/10.3389/fimmu.2012.00051
[41] Sawant, D. V., Hamilton, K., & Vignali, D. A. (2015). Interleukin-35: Expanding Its Job Profile. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research, 35(7), 499–512. https://doi.org/10.1089/jir.2015.0015
[42] Cedeno-Laurent, F., Opperman, M., Barthel, S. R., Kuchroo, V. K., & Dimitroff, C. J. (2012). Galectin-1 triggers an immunoregulatory signature in Th cells functionally defined by IL-10 expression. Journal of immunology (Baltimore, Md. : 1950), 188(7), 3127–3137. https://doi.org/10.4049/jimmunol.1103433
[43] Cedeno-Laurent, F., Opperman, M., Barthel, S. R., Kuchroo, V. K. & Dimitroff, C. J. Galectin-1 triggers an immunoregulatory signature in T helper cells functionally defined by IL-10 expression. J. Immunol. Baltim. Md 1950 ,2012, 188, 3127–3137.
[44] Whiteside, T. L. & Jackson, E. K. Adenosine and Prostaglandin E2 Production by Human Inducible Regulatory T Cells in Health and Disease. Front. Immunol. 2013, 4.
[45] Mandapathil, M. et al. Increased ectonucleotidase expression and activity in Treg of patients with head and neck cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 6348–6357.
[46] Levring, T. B. et al. Activated human CD4+ T cells express transporters for both cysteine and cystine. Sci. Rep. 2012,2.
[47] Yan, Z., Garg, S. K. & Banerjee, R. Regulatory T Cells Interfere with Glutathione Metabolism in Dendritic Cells and T Cells. J. Biol. Chem. 2010,285, 41525–41532.
[48] Liu, X., Mo, W., Ye, J., Li, L., Zhang, Y., Hsueh, E. C., Hoft, D. F., & Peng, G.. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nature communications, 2018, 9(1), 249. https://doi.org/10.1038/s41467-017-02689-5
[49] McGeachy, M. J., & Cua, D. J.. Th17 cell differentiation: the long and winding road. Immunity, 28(4), 445–453. https://doi.org/10.1016/j.immuni.2008.03.001
[50] Brembilla, N. C., Senra, L. & Boehncke, W.-H. The IL-17 Family of Cytokines in Psoriasis: IL-17A and Beyond. Front. Immunol. 2018,9.
[51] Zhu, J. & Paul, W. E. Peripheral CD4 T cell differentiation regulated by networks of cytokines and transcription factors. Immunol. Rev. 2010, 238, 247–262.
[52] Tsai, H.-C., Velichko, S., Hung, L.-Y. & Wu, R. IL-17A and Th17 Cells in Lung Inflammation: An Update on the Role of Th17 Cell Differentiation and IL-17R Signaling in Host Defense against Infection. Clinical and Developmental Immunology vol. 2013 e267971 https://www.hindawi.com/journals/jir/2013/267971/ (2013).
[53] Waite, J. C. & Skokos, D. Th17 Response and Inflammatory Autoimmune Diseases. International Journal of Inflammation vol. 2012 e819467 https://www.hindawi.com/journals/iji/2012/819467/ (2011).
[54] Chen, X. & Oppenheim, J. J. Th17 cells and Tregs: unlikely allies. J. Leukoc. Biol. 2014,95, 723–731.
[55] Du, R., Zhao, H., Yan, F. & Li, H. IL-17+Foxp3+ T cells: an intermediate differentiation stage between Th17 cells and regulatory T cells. J. Leukoc. Biol. 2014,96, 39–48.
[56] Ye, J., Livergood, R. S. & Peng, G. The Role and Regulation of Human Th17 Cells in Tumor Immunity. Am. J. Pathol. 2013, 182, 10–20.
[57] Stadhouders, R., Lubberts, E. & Hendriks, R. W. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J. Autoimmun. 2018, 87, 1–15.
[58] Martin, F., Apetoh, L. & Ghiringhelli, F. Controversies on the role of Th17 in cancer: a TGF-β-dependent immunosuppressive activity? Trends Mol. Med. 2012, 18, 742–749.
[59] Lee, G. R. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19.
[60] Knochelmann, H. M. et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell. Mol. Immunol. 2018, 15, 458–469.
[61] Duan, M. C., Zhong, X. N., Liu, G. N., & Wei, J. R. (2014). The Treg/Th17 paradigm in lung cancer. Journal of immunology research, 2014, 730380. https://doi.org/10.1155/2014/730380
[62] De Simone, V., Pallone, F., Monteleone, G. & Stolfi, C. Role of TH17 cytokines in the control of colorectal cancer. Oncoimmunology 2013, 2.
[63] Martiniuk, F. et al. TH17 is involved in the remarkable regression of metastatic malignant melanoma to topical diphencyprone. J. Drugs Dermatol. JDD 2010, 9, 1368–1372.
[64] Lippens, C., Duraes, F. V., Dubrot, J., Brighouse, D., Lacroix, M., Irla, M., Aubry-Lachainaye, J. P., Reith, W., Mandl, J. N., & Hugues, S. (2016). IDO-orchestrated crosstalk between pDCs and Tregs inhibits autoimmunity. Journal of autoimmunity, 75, 39–49. https://doi.org/10.1016/j.jaut.2016.07.004
[65] Guéry, L. & Hugues, S. Th17 Cell Plasticity and Functions in Cancer Immunity. BioMed Research International vol. 2015 e314620 https://www.hindawi.com/journals/bmri/2015/314620/ (2015).
[66] Chatterjee, S., Thyagarajan, K., Kesarwani, P., Song, J. H., Soloshchenko, M., Fu, J., Bailey, S. R., Vasu, C., Kraft, A. S., Paulos, C. M., Yu, X. Z., & Mehrotra, S. (2014). Reducing CD73 expression by IL1β-Programmed Th17 cells improves immunotherapeutic control of tumors. Cancer research, 74(21), 6048–6059. https://doi.org/10.1158/0008-5472.CAN-14-1450
[67] Hayata, K. et al. Inhibition of IL-17A in Tumor Microenvironment Augments Cytotoxicity of Tumor-Infiltrating Lymphocytes in Tumor-Bearing Mice. PLoS ONE ,2013, 8.
[68] Alizadeh, D., Katsanis, E. & Larmonier, N. The Multifaceted Role of Th17 Lymphocytes and Their Associated Cytokines in Cancer. Clin. Dev. Immunol. 2013.10. 1-11.
[69] Wilke, C. M., Kryczek, I., Wei, S., Zhao, E., Wu, K., Wang, G., & Zou, W. (2011). Th17 cells in cancer: help or hindrance? Carcinogenesis, 32(5), 643–649. https://doi.org/10.1093/carcin/bgr019
[70] Karin, N. CXCR3 Ligands in Cancer and Autoimmunity, Chemoattraction of Effector T Cells, and Beyond. Front. Immunol. 2020,11.
[71] Guéry, L. & Hugues, S. Th17 Cell Plasticity and Functions in Cancer Immunity. BioMed Research International vol. 2015 e314620 https://www.hindawi.com/journals/bmri/2015/314620/ (2015).
[72] Schwartz, R. H. T cell anergy. Annu. Rev. Immunol. 2003, 21, 305–334.
[73] Lechler, R., Chai, J.-G., Marelli-Berg, F. & Lombardi, G. The contributions of T-cell anergy to peripheral T-cell tolerance. Immunology 2001, 103, 262–269.
[74] Nurieva, R., Wang, J., & Sahoo, A. (2013). T-cell tolerance in cancer. Immunotherapy, 5(5), 513–531. https://doi.org/10.2217/imt.13.33
[75] Jung, K. & Choi, I. Emerging Co-signaling Networks in T Cell Immune Regulation. Immune Netw. 2013,13, 184–193.
[76] Wherry, E. J. & Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499.
[77] Riley, J. L. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125.
[78] Zhao, Y., Shao, Q. & Peng, G. Exhaustion and senescence: two crucial dysfunctional states of T cells in the tumor microenvironment. Cell. Mol. Immunol. 2020, 17, 27–35.
[79] Li, L., Wan, S., Tao, K., Wang, G. & Zhao, E. KLRG1 restricts memory T cell antitumor immunity. Oncotarget 2016, 7, 61670–61678.
[80] Li, H. et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351.
[81] Gattinoni L.. Memory T cells officially join the stem cell club. Immunity, 2014, 41(1), 7–9. https://doi.org/10.1016/j.immuni.2014.07.003
[82] Pennock, N. D. et al. T cell responses: naïve to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283.
[83] Steinbach, K., Vincenti, I. & Merkler, D. Resident-Memory T Cells in Tissue-Restricted Immune Responses: For Better or Worse? Front. Immunol. 2018, 9.
[84] Samji, T. & Khanna, K. M. Understanding Memory CD8+ T cells. Immunol. Lett. 2017, 185, 32–39.
[85] Kagoya, Y., Nakatsugawa, M., Ochi, T., Cen, Y., Guo, T., Anczurowski, M., Saso, K., Butler, M. O., & Hirano, N. (2017). Transient stimulation expands superior antitumor T cells for adoptive therapy. JCI insight, 2(2), e89580. https://doi.org/10.1172/jci.insight.89580
[86] Tanel, A., Fonseca, S. G., Yassine-Diab, B., Bordi, R., Zeidan, J., Shi, Y., Benne, C., & Sékaly, R. P. (2009). Cellular and molecular mechanisms of memory T-cell survival. Expert review of vaccines, 8(3), 299–312. https://doi.org/10.1586/14760584.8.3.299
[87] Muranski, P., Borman, Z. A., Kerkar, S. P., Klebanoff, C. A., Ji, Y., Sanchez-Perez, L., Sukumar, M., Reger, R. N., Yu, Z., Kern, S. J., Roychoudhuri, R., Ferreyra, G. A., Shen, W., Durum, S. K., Feigenbaum, L., Palmer, D. C., Antony, P. A., Chan, C. C., Laurence, A., Danner, R. L., … Restifo, N. P. . Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity, 2011, 35(6), 972–985. https://doi.org/10.1016/j.immuni.2011.09.019
[88] Wei, S., Zhao, E., Kryczek, I. & Zou, W. Th17 cells have stem cell-like features and promote long-term immunity. Oncoimmunology 2012, 1, 516–519.
[89] Zhu, J., Yamane, H., & Paul, W. E.. Differentiation of effector CD4 T cell populations (*). Annual review of immunology, 2010, 28, 445–489. https://doi.org/10.1146/annurev-immunol-030409-101212
[90] De Groot, R., Van Loenen, M. M., Guislain, A., Nicolet, B. P., Freen-Van Heeren, J. J., Verhagen, O., Van Den Heuvel, M. M., De Jong, J., Burger, P., Van Der Schoot, C. E., Spaapen, R. M., Amsen, D., Haanen, J., Monkhorst, K., Hartemink, K. J., & Wolkers, M. C. (2019). Polyfunctional tumor-reactive T cells are effectively expanded from non-small cell lung cancers, and correlate with an immune-engaged T cell profile. Oncoimmunology, 8(11), e1648170. https://doi.org/10.1080/2162402X.2019.1648170
[91] Burel, J. G., Apte, S. H., Groves, P. L., McCarthy, J. S., & Doolan, D. L. (2017). Polyfunctional and IFN-γ monofunctional human CD4+ T cell populations are molecularly distinct. JCI insight, 2(3), e87499. https://doi.org/10.1172/jci.insight.87499
[92] Soghoian, D. Z., & Streeck, H. (2010). Cytolytic CD4(+) T cells in viral immunity. Expert review of vaccines, 9(12), 1453–1463. https://doi.org/10.1586/erv.10.132
[93] Salido, J., Ruiz, M. J., Trifone, C., Figueroa, M. I., Caruso, M. P., Gherardi, M. M., Sued, O., Salomón, H., Laufer, N., Ghiglione, Y., & Turk, G. (2018). Phenotype, Polyfunctionality, and Antiviral Activity of in vitro Stimulated CD8+ T-Cells From HIV+ Subjects Who Initiated cART at Different Time-Points After Acute Infection. Frontiers in immunology, 9, 2443. https://doi.org/10.3389/fimmu.2018.02443
[94] Bhat, P., Leggatt, G., Waterhouse, N., & Frazer, I. H. (2017). Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell death & disease, 8(6), e2836. https://doi.org/10.1038/cddis.2017.67
[95] Ding, Z. C., Huang, L., Blazar, B. R., Yagita, H., Mellor, A. L., Munn, D. H., & Zhou, G. (2012). Polyfunctional CD4? T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood, 120(11), 2229–2239. https://doi.org/10.1182/blood-2011-12-398321
[96] Seidel, J. A., Otsuka, A., & Kabashima, K. (2018). Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Frontiers in oncology, 8, 86. https://doi.org/10.3389/fonc.2018.00086
[97] Gallimore, A., & Godkin, A. (2008). Regulatory T cells and tumour immunity - observations in mice and men. Immunology, 123(2), 157–163. https://doi.org/10.1111/j.1365-2567.2007.02748.
[98] Kim, J. H., Kim, B. S., & Lee, S. K. (2020). Regulatory T Cells in Tumor Microenvironment and Approach for Anticancer Immunotherapy. Immune network, 20(1), e4. https://doi.org/10.4110/in.2020.20.e4
[99] Toor, S. M., Murshed, K., Al-Dhaheri, M., Khawar, M., Abu Nada, M., & Elkord, E. (2019). Immune Checkpoints in Circulating and Tumor-Infiltrating CD4+ T Cell Subsets in Colorectal Cancer Patients. Frontiers in immunology, 10, 2936. https://doi.org/10.3389/fimmu.2019.02936
[100] Ohue, Y. & Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089.
[101] Han, S., Toker, A., Liu, Z. Q. & Ohashi, P. S. Turning the Tide Against Regulatory T Cells. Front. Oncol. 2019, 9.
[102] Dai, H., Wang, Y., Lu, X. & Han, W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. JNCI J. Natl. Cancer Inst. 2016, 108.

- Trends in Oncology的其它文章
- Role of Cetuximab Re-introduction and Re-challenge in Later Lines of Treatment in Metastatic Colorectal Cancer: A Case Series
- To Use or not to Use Anticoagulation in Patients with Advanced Malignancies? – This is the Question
- A Preliminary Phase-2 Study with very High-Dose of Melatonin (1000 mg/day) in Untreatable Advanced Cancer Patients Already Progression on Previous Palliative Therapy with High-Dose Melatonin
- Special Interview