
LC-MS/MS法测定人血浆中曲格列汀的浓度及2种片剂药动学比较研究
2020-08-28 05:43谢朋飞崔宏娣陈元璐龙辉金玉任军李雪芳杨鹏周燕张永东
广东药科大学学报 2020年4期
谢朋飞,崔宏娣,陈元璐,龙辉,金玉,任军,李雪芳,杨鹏,周燕,张永东
(1.郴州市第一人民医院Ⅰ期临床研究室,湖南 郴州 423000; 2.上海海虹实业(集团)巢湖今辰药业有限公司,安徽 巢湖 238000; 3.安徽万邦医药科技股份有限公司,安徽 合肥 230601)
曲格列汀(Trelagliptin,TRE)是由日本武田药品工业株式会社和Furiex公司共同开发的全球首个长效二肽基肽酶IV(DPP-4)抑制剂,通过选择性、持续性抑制DPP-4而控制血糖水平[1-3]。曲格列汀具有良好的安全性和耐受性,每周给药1次便可有效控制血糖水平,患者用药依从性好,市场前景广阔[4-5]。
目前,文献报道曲格列汀检测方法有HPLC法、LC-MS/MS法[6-10],但对于生物样品中曲格列汀的检测方法报道较少。本研究旨在建立一种具有高灵敏度和高选择性的HPLC-MS/MS法测定人血浆中曲格列汀的浓度,计算空腹和餐后给药条件下琥珀酸曲格列汀的药动学特征,初步评价受试制剂和参比制剂(商品名:Zafatekl®)的生物等效性,为正式试验和处方工艺筛选优化提供参考依据。
1 材料与方法
1.1 药品与试剂
受试制剂(T):琥珀酸曲格列汀片(巢湖今辰药业有限公司,规格:100 mg/片,批号:171205);参比制剂(R):琥珀酸曲格列汀片(商品名:Zafatekl®,日本武田药品工业株式会社,规格:100 mg/片,批号:17N0010);琥珀酸曲格列汀对照品(济南瑞丰医药科技有限公司,质量分数99.6%,批号:161114);内标(卡马西平Carbamazepine,CAR,质量分数99.7%,批号:100142-201105,中国食品药品检定研究院);甲醇、乙腈、异丙醇、甲酸(Sigma公司);屈臣氏饮用水。
1.2 仪器
岛津LC-30A液相色谱仪(日本Shimadzu公司);AB QTRAP 5500质谱仪(ESI离子源,美国AB公司);Analyst 1.6.3数据处理软件;XP6百万分之一电子天平(梅特勒-托利多仪器(上海)有限公司)。
1.3 试验对象
经郴州市第一人民医院药物临床试验伦理委员会批准,所有受试者均在签署书面知情同意书后进入试验。空腹试验入选8例受试者,其中男性3例,女性5例,年龄21~46 a,体重指数19.9~26.0 kg/m2;餐后试验入选8例受试者,其中男性5例,女性3例,年龄20~40 a,体重指数20.5~25.1 kg/m2。
入选标准:年龄18~65 a,男女均可,健康情况良好;体重指数19.0~26.0 kg/m2,男性受试者体重≥50.0 kg,女性受试者体重≥45.0 kg。试验前签署知情同意书、并对试验内容、过程及可能出现的不良反应充分了解;且能够按照试验方案要求完成研究。
排除标准:生命体征、体格检查及血常规、尿常规、血生化、输血四项、妊娠检查(女性)、12导联心电图、腹部B超(肝、胆、脾、肾)、药物滥用筛查、酒精呼气等检查异常且具有临床意义者;有心、肝、肾、内分泌、消化道、免疫系统、呼吸系统及神经系统等严重疾病史或现有上述系统疾病者。
1.4 研究设计
1.4.1 总体设计 单中心、空腹/餐后、随机、开放、单剂量、两序列两周期双交叉设计,空腹8例,餐后8例,清洗期18 d。受试人群:普通人群,男女均有,性别比例适当。随机分组方法:受试者按筛选号顺序1∶1比例随机分配到2个给药顺序组(T-R组/R-T组)之一。给药剂量:100 mg。给药方法:空腹或者高脂餐后30 min口服,240 mL温水送服。服药前禁食至少10 h,服药前1 h及服药后1 h内禁止饮水,服药后4 h内禁食,服药后4、10 h统一进食标准午餐和晚餐。
1.4.2 样本采集与检测 空腹及餐后采血时间点:给药前0 h(服药前1 h内)和给药后0.25、0.5、0.75、1.0、1.25、1.5、1.75、2.0、2.5、3.0、4.0、6.0、8.0、12.0、24.0、48.0、72.0、96.0、120.0、144.0、168.0 h;每次采血约4 mL置于EDTA-K2抗凝剂真空负压管中,轻柔颠倒混匀4~8次,采集后即刻置于冰浴中,于1 h内在2~8 ℃离心10 min(离心力:2 500 g),分离出血浆,2 h内放置超低温冰箱(-70±10)℃冻存,直至样本转运。转运过程全程冷链监控(约-80 ℃)。采用HPLC-MS/MS法测定受试者口服受试制剂或参比制剂后血浆中曲格列汀的血药浓度。
1.4.3 血浆样本处理与测定条件
1.4.3.1 色谱条件 色谱柱为Welth Ultimate XB-C18(5 μm,2.1 mm×100 mm);0.1%甲酸为流动相A,乙腈为流动相B,梯度洗脱(0~0.5 min,95%A;1.2 min,35%A;2.8 min,5%A;3 min,95%A);流速:0.5 mL/min,梯度洗脱;柱温30.0 ℃,进样器温度为4 ℃,进样量5 μL。
1.4.3.2 质谱条件 质谱条件采用正离子电喷雾离子化(ESI)电离模式,多反应监测模式(MRM)扫描模式。曲格列汀离子对m/z358.2→134.0,内标卡马西平离子对m/z237.1→194.0。离子源参数设置:喷雾电压为5.5 kV,气帘气241.33 kPa,雾化气为344.75 kPa,辅助加热气为379.23 kPa,离子源温度550 ℃,入口电压为10 V。
1.4.3.3 标准曲线、质控血浆样品制备 精密称取适量的曲格列汀对照品,用甲醇溶液配制曲格列汀储备液并稀释制得标准曲线系列工作液,同法配制质控(QC)储备液及工作液。精密称取适量的卡马西平对照品,用甲醇溶液配制卡马西平储备液并稀释成10 ng/mL内标工作溶液。临用前以标准曲线工作液10 μL加入空白血浆190 μL的比例混匀即得标准曲线血浆样品。标准曲线血浆样品STD1~STD8(含曲格列汀1、2、5、20、80、320、640、800 ng/mL)。各质控点浓度值为:LLOQ(1 ng/mL)、LQC(3 ng/mL)、M1QC(25 ng/mL)、M2QC(200 ng/mL)、HQC(600 ng/mL)。每批随行标准曲线均合格(各浓度点的回算浓度与理论浓度的偏差在±15%范围内,定量下限的偏差在±20%范围内;包含至少6个符合接受标准的非零浓度点;回归方程的线性相关系数r>0.99)。
1.4.3.4 血浆样品处理 在96孔板中加入50 μL样品;双空白样品,加入体积分数50%甲醇50 μL,其他样品加入内标工作液50 μL,涡旋混匀;每一个样品孔中加入乙腈200 μL,封板,混匀5 min;样品在4 ℃、4 200 r/min离心10 min;离心后转移100 μL上清液至另一96孔板中,加入200 μL纯水,封板,混匀5 min;样品在4 ℃、 4 200 r/min离心10 min,取上清液进样。
1.5 方法学考察与评价
1.5.1 专属性考察 取6份不同批次未混合的健康受试者空白EDTA-K2血浆,分别进行不加内标的空白样品测定和在LLOQ水平的添加试验。结果显示,空白人血浆样本在待测物出峰处的峰面积不超过定量下限待测物峰面积的20.0%,内标物出峰处的峰面积不超过定量下限样品的内标物峰面积的5.0%,表明方法专属性良好,血浆中内源性物质不干扰待测物和内标的测定,见图1。
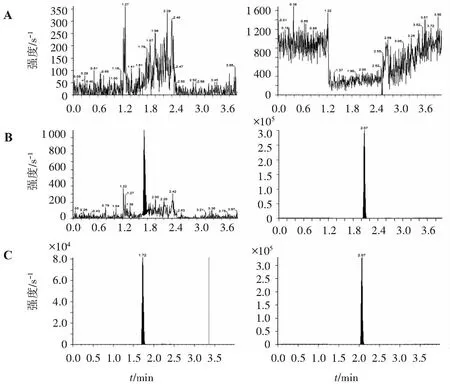
A.空白基质; B.定量下限LLOQ样品(曲格列汀:1 ng/mL)和内标卡马西平; C.标准曲线工作液STD05样品(曲格列汀:80 ng/mL)和内标卡马西平; 曲格列汀(tR=1.72 min); 卡马西平(tR=2.07 min)。
1.5.2 标准曲线与定量下限 配制标准曲线血浆样品STD1~STD8(含曲格列汀1、2、5、20、80、320、640、800 ng/mL),按“血浆样本处理”项下操作后进样分析,建立曲格列汀的标准曲线。以待测物曲格列汀浓度(X,ng/mL)为横坐标,待测物色谱峰面积(As)与内标色谱峰面积(Ai)的比值(Y=As/Ai)为纵坐标,用加权(1/X2)最小二乘法进行线性回归运算。结果显示,曲格列汀在1~800 ng/mL内线性关系良好,标准曲线为Y=3.32×10-3X+2.21×10-4(R2= 0.992),最低定量下限为1 ng/mL(LLOQ)。
1.5.3 精密度与回收率试验 量取曲格列汀LLOQ、LQC、M1QC、M2QC和HQC 5个质量浓度的质控样品考察批内及批间精密度与准确度。每一浓度6份,连续测定3个批次。按“血浆样本处理”项下操作后进样分析,以测得浓度计算日内及日间精密度。用低(LQC)、中(M2QC)、高(HQC)3个质量浓度水平的样本计算人血浆提取样本加内标的峰面积比值,与空白人血浆经提取后的样本再加入代谢物和内标后测得的峰面积比值进行比较,计算回收率。结果5个质量浓度水平的质控样品检测结果的批内和批间精密度均小于15.0%,回收率在92.9%~97.6%之间,见表1。
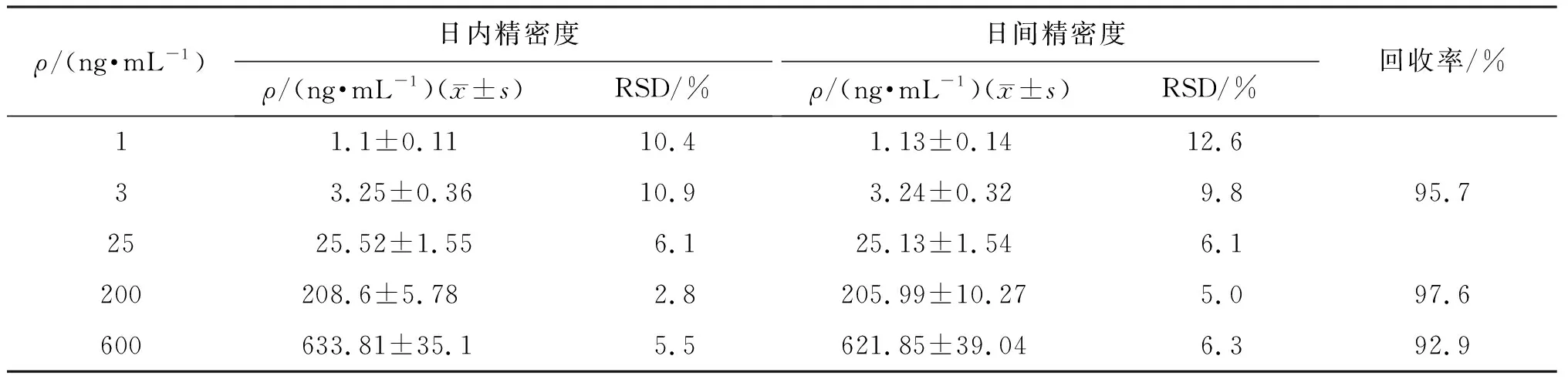
表1 曲格列汀在人血浆中的精密度和回收率Table 1 Precision and recovery of trelagliptin in human plasma
1.5.4 基质效应 用6份不同来源的空白基质考察内标工作浓度下和待测物低、中、高(3、200、600 ng/mL)3个质量浓度水平下的基质效应。3个质量浓度水平内标归一化的基质因子值为98.8%~101.8%,精密度≤5.3%,符合要求。分别用模拟的溶血血浆(在正常的空白血浆中加入一定量溶血的全血得到2%溶血水平的溶血血浆)及高脂血浆[在正常空白血浆加入脂肪乳注射液(3 mg/mL,以甘油三酯含量计算)制备低浓度(LQC 3 ng/mL)和高浓度(HQC 600 ng/mL)]的质控样品,每个浓度平行处理6份样品。溶血基质中LQC、HQC基质效应的CV值分别为7.1%、0.8%;高脂基质中LQC、HQC基质效应的CV值分别为5.9%、3.9%,均≤15%,表明溶血基质及高脂血基质均不影响血浆中曲格列汀的准确定量,符合生物样品分析要求。
1.5.5 稳定性试验 配制含曲格列汀的低、高质量浓度(3、600 ng/mL)的全血和血浆质控样本,分别考察全血质控样本室温放置2 h、血浆质控样本室温下立即处理、室温放置29 h、-20 ℃/-70 ℃反复冻融5次、-20 ℃/-70 ℃长期存放(至少涵盖从第1个样品采集至样品分析结束的时间段)、样本处理后置于室温条件下5 h、自动进样器中或与自动进样器相同温度的环境77 h后的稳定性。结果表明,低、高质量浓度全血和血浆质控样本在上述条件下均稳定性良好。
1.6 药动学参数计算及生物等效性判定
使用非房室模型(NCA)方法,用WinNonlin 7.0软件进行统计分析,计算药动学参数,对主要药动学参数(Cmax、AUC0-t、AUC0-∞)进行自然对数转换后,考虑周期效应、制剂效应、序列效应、受试者(随序列)效应和残差,以lnCmax、lnAUC0-t、lnAUC0-∞为因变量进行多因素方差分析,对Tmax进行非参数检验(Wilcoxon符号秩检验)。以方差分析结果为基础,采用90%CI分析法,当受试制剂与参比制剂药动学参数AUC0-t、AUC0-∞和Cmax的几何均值比的90%CI均在80.00%~125.00%等效区间内,则认为两制剂生物等效。通过观察生命体征、实验室检测及心电图等不良事件进行安全性评价。
2 结果
2.1 平均血药浓度-时间曲线
空腹试验8例受试者、餐后试验8例受试者完成2周期琥珀酸曲格列汀片(100 mg)受试制剂和参比制剂给药,按方案要求在规定时间内完成血样采集,并进行血药浓度检测。平均血药浓度-时间曲线见图2、图3。
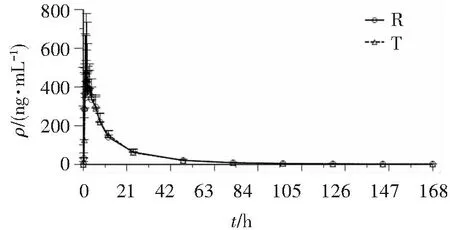
图2 空腹组受试者单次口服琥珀酸曲格列汀片受试制剂(T)和参比制剂(R)后平均血药浓度-时间曲线Figure 2 Mean plasma concentration-time curve of TRE after taking the test and reference drugs in fasting state (n=8)
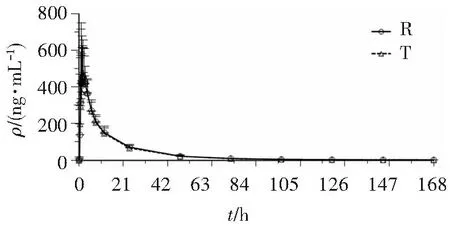
图3 餐后组受试者单次口服琥珀酸曲格列汀片受试制剂(T)和参比制剂(R)后平均血药浓度-时间曲线Figure 3 Mean plasma concentration-time curve of TRE after taking the test and reference drugs in fed state (n=8)
2.2 药动学参数
本临床试验过程控制良好,两周期间基本保持一致,入组受试者均完成试验,无脱落,未发生严重不良事件。空腹试验组8例受试者和餐后试验组8例受试者均纳入药动学参数集(PKPS)分析,健康受试者空腹和餐后单次口服受试药物和参比药物后血浆中琥珀酸曲格列汀片的药动学参数见表2。
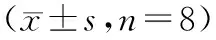
表2 空腹组和餐后组受试者单次口服琥珀酸曲格列汀片受试制剂(T)和参比制剂(R)的药动学参数Table 2 The main pharmacokinetic parameters of TRE after taking the test or reference drugs in fasting and fed state
2.3 生物等效性评价
在空腹和餐后给药条件下受试制剂和参比制剂主要药动学参数lnAUC0-t及lnAUC0-∞的几何均数比值90%置信区间均在80.00%~125.00%范围内,空腹及餐后给药条件下lnCmax几何均数比值的90%置信区间上限超出了可接受80.00%~125.00%等效范围内,并经双单侧t-检验分析后,高侧P值>0.05,则接受H0拒绝H1,即两制剂在本试验餐后给药条件下Cmax不具有生物等效性,见表3。

表3 空腹组和餐后组受试者单次口服琥珀酸曲格列汀片受试制剂(T)和参比制剂(R)的90%置信区间 (n=8)Table 3 The 90% CIs for the main pharmacokinetic parameters of TRE after taking the test or reference drugs in fasting and fed state
2.4 安全性评价
空腹试验8例受试者全部按计划完成给药、采血和安全性评价,并进入安全性分析集。共2例受试者发生2例次不良事件,均系服用受试制剂发生不良事件。不良事件为高尿酸血症、肠炎,严重程度均为Ⅰ级(NCI CTCAE4.03),未进行任何治疗,均恢复正常。
餐后试验8例受试者全部按计划完成给药、采血和安全性评价,并进入安全性分析集。共4例受试者发生5例次不良事件,其中服用受试制剂的8例受试者中发生2例次不良事件;服用参比制剂的8例受试者中发生3例次不良事件。不良事件为口腔黏膜炎、尿路感染、黄疸、肠炎,所有不良事件严重程度均为Ⅰ级,未进行任何治疗,均恢复正常。
本研究无严重不良事件,无受试者因不良事件退出试验,琥珀酸曲格列汀片(100 mg)受试制剂及参比制剂均具有较好的安全性。
3 讨论
本研究建立了健康人血浆中曲格列汀LC-MS/MS测定方法。参照CFDA《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》以及琥珀酸曲格列汀片原研说明书,确定生物样品测定的目标待测物为曲格列汀。生物样本前处理采用乙腈蛋白沉淀法,操作简单,耗时短,适用于高通量样本检测;采用卡马西平作为内标,价格便宜、易得,且与待测物曲格列汀有相似的理化性质,可以有效减少基质效应、回收率等导致检测结果不可靠的情况。采用LC-MS/MS(ESI+离子化,MRM模式)方法,具有选择性强,分析时间快,自动化程度高,检测限低等优点。经方法学验证本法符合2015年版《中国药典》中“生物样品定量分析方法验证指导原则”的相关要求,可用于临床试验血浆样品的分析检测。
曲格列汀为生物药剂学分类系统第三类药物(BCS Ⅲ类:高溶解性,低渗透性),药物渗透性是这类药物吸收的限速步骤,且胃肠道滞留时间和管腔组成以及膜渗透性的个体差异均会影响药物在体内吸收[11-12]。研究显示,餐后给药相比于空腹给药,受试制剂Cmax减少了2.98%,AUC0-t和AUC0-∞没有明显变化;参比制剂Cmax没有明显变化,AUC0-t和AUC0-∞分别增加了5.04%、5.36%,空腹和餐后各药动学参数个体内变异相似。本研究结果显示,巢湖今辰药业有限公司生产的琥珀酸曲格列汀片空腹和餐后给药条件下吸收速度和程度与参比制剂基本一致,主要药动学参数与参比制剂说明书报道相近。本研究中空腹及餐后采血点在3.5 h均有分布,建议在正式试验时增加3.5 h采样点;截取72 h后分析的结果,AUCt/AUCi已超过95%,正式试验可设计采血终点为72 h 。综上所述,建议正式试验采血点可设计为给药前0 h及给药后0.25、0.5、0.75、1.0、1.25、1.5、1.75、2.0、2.5、3.0、3.5、4.0、6.0、8.0、12.0、24.0、48.0、72.0 h,共计19个采样点。本研究中未参考FDA关于降糖药服药指南使用葡萄糖水送服药物,未出现低血糖反应,因此正式试验仍可考虑不用葡萄糖水送服。本研究中实际测得Cmax的CV值较大,在8例受试者的样本量下无法满足等效结果,根据本研究空腹/餐后受试者中的Cmax个体内变异(Intra-subject CV)系数、θ值以及实际临床过程中考虑20%脱落率,采用 PASS11.0 样本量统计软件预估满足生物等效条件所需的最低试验样本量空腹为28例、餐后为30例。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
口腔护理用品工业(2021年4期)2021-11-02
中成药(2019年12期)2020-01-04
中国酿造(2018年9期)2018-11-05
中成药(2018年6期)2018-07-11
浙江工业大学学报(2017年5期)2018-01-22
中成药(2017年10期)2017-11-16
中成药(2017年5期)2017-06-13
中国粮油学报(2016年5期)2016-01-23
中国洗涤用品工业(2015年9期)2015-02-28
