
普鲁兰酶基因在毕赤酵母中的表达研究
2020-08-27 12:58王颢霖
河南农业·教育版 2020年7期
关键词:克隆
王颢霖

关键词:普鲁兰酶;克隆;毕赤酵母;诱导表达
在淀粉的加工行业中,高压酸解法作为葡萄糖生产的老工艺具有需要设备少、耗时短的优点,但其副产物较多,耗费的酸和碱的量较高,而且为了去除苦味必须采用严格控制温度、缓慢结晶的办法,需要淀粉加工工厂投入较高的人力资源和设备成本;如果运用a-淀粉酶作用于较长的淀粉分子使其断裂成小分子,使高浓度的淀粉糊在高温下糊化液化,再运用糖化型淀粉酶,就可以高效地得到目的产物。此法具有生产率高、成本低、副产物少、结晶快等优点,对于生产实践具有很大的意义。
普鲁兰酶最早是在肺炎克雷伯氏菌中被发现,并且被发现可以水解支链淀粉中的a-1,6糖苷键,不仅能够极大提高淀粉的水解效率,还能得到具有高特异性的产物,对于淀粉的糖化步骤十分重要。自然界中的天然酶一般都具有含量低、提取较难、不易制备的弊端,通常在自然界中筛选特定性质酶的效率很低。而研究酶在工程菌株中的克隆与表达,选育出更好的产酶菌株,对于获得更高酶活性,更高表达量的性能优良的酶具有非常重要的意义。
各项研究表明,普鲁兰酶可被许多菌种表达,如缓和链球菌,大肠杆菌,耐热产硫梭菌,枯草芽孢杆菌,酵母菌等等。在原核表达系统中,大肠杆菌表达系统作为研究较为深入的表达系统,在1984年最早成功异源表达了普鲁兰酶,但是其翻译后修饰的过程比较简单,表达量也很低。经过科研人员的不断努力,对表达宿主大肠杆菌进行优化和对发酵条件进行改善,生产出的酶活力曾经高达502U/mL,具有很强的工业应用前景。但是其高密度发酵时的温度一般是低于37℃的,不利于细菌的快速生长;枯草芽孢杆菌也具有清晰的遗传背景和生理特性,未经优化的情况下,表达普鲁兰酶的活力可达到10.94U/mL。两者都具有一定的工业生产普鲁兰酶的潜力。
真核生物Pichia pastoffs最早是在1987年被Cregg研究将其作为宿主成功的进行了外源蛋白的表达实验,被发现其在外源表达方面所具有的潜力。随着人们对Piehia pas-toffs研究的深入,发现Pichiapastoffs在外源表达方面的独特优势,与其他表达系统比较起来的优点大体上为:对细胞没有毒害,是一种安全的表达宿主。表达的量通常较高,可以获得大量的目的蛋白,部分蛋白的表达量高至12g/L。不但可以用于分泌型表达,还可以用于细胞内表达。表达出的蛋白易于分离纯化。某些在细菌系统中表达不好的真核基因,在酵母系统中表达情况良好。能够在廉价的培养基上生长,十分节省实验成本,方便用于外源基因的操作。作为真核生物,其转录翻译后加工、外分泌、翻译后修饰以及糖基化修饰的性能良好,可以使表达产物更加接近于天然蛋白质,甚至与天然蛋白质一样。利用高表达的启动子MOX、AOX、LAC4等,外源基因可同源重组到Pichia pastoris的基因组中进行稳定复制,有利于外源基因的表达。曾用于生产SCP,有着清晰的遗传背景和良好的发酵基础。细胞生长速度快,适用于高密度发酵能够移去起始Met,使重组蛋白在用于临床应用时不引起免疫反应。因此,对医疗行业起到很大的贡献。
实验前期将来自于本实验室的普鲁兰酶基因在大肠杆菌BL21 DE3中进行表达,其表达量非常低。换用能缓解大肠杆菌稀有密码子问题大肠杆菌Rossta DE3也未能明显提高蛋白的表达量。
本实验旨在通过以更换表达宿主的方式提高蛋白的表达量。即将目的基因同源重组入PichiapastorisGSll5,对重组菌进行诱导表达,分析其蛋白产率。
一、材料与方法
(一)材料
1、菌株和质粒。含有Thermogota thermarum中获取的普鲁兰酶基因的重组质粒pET21a-TT由河南农业大学推广楼酶工程504实验室构建和保存,毕赤酵母(Piehia pasto-ris P.pastoris)GSll5和表达载体pPIC3.5K由河南农业大学酶工程实验室保存。
2、工具酶和主要试剂。山梨醇(D-Sorbit01),琼脂(Agar Powder),酵母提取物(Yeast Extract),质粒小提试剂盒,PCR产物回收试剂盒,胶回收DNA试剂盒,椰油醇硫酸钠(SDS)胰蛋白胨(Ttyton),琼脂糖(Agarose),三羟基甲基氨基甲烷(riffs),氨苄青霉素(Ampicillin),甘氨酸(Glycine),三羟甲基氨基甲烷(Tris),乙二胺四乙酸(EDTA)等。甲醇,异丙醇等普通试剂均为分析纯试剂。各种工具酶购自SMOBIL公司。设计引物如下:
TF-EcoRI-F:CCGGAATFCGCCACCATGAAAAGGCTAC-337TACTCATCGTCATAACTTGTACAG
Tr-NotI-R:ATAAGAATGCGGCCGCTCAGTGGTGGTGG-TGGTGG
5"AOX GACTGGTTCCAATYGACAAGC;
3"AOX GCAAATGGCATTCTGACATCC
引物交由苏州金唯智生物科技有限公司合成。目的基因和载体测序以及转化子测序由郑州何泽公司完成。
(二)方法
1、含目的基因载体的验证。从-80℃冰箱取出10%甘油菌保存的重组菌划线于带氨苄抗性的LB平板,在恒温培养箱37℃培养活化12~16h。挑取单菌落于5mL带氨苄抗性LB培养试管中,放置于摇床37℃,220 rpm培养12~16h。提取质粒。测定浓度并进行琼脂糖凝胶电泳,将位置正确的质粒送去测序公司进行测序。
2、重组质粒的构建。使用引物TT-EcoRI-F與TT-No.tI-R扩增目的基因片段。按如下体系将样品加入PCR管:ddH20 38 txL;TT-EcoRI-F 2.5μL;TY-Nod-R 2.5μL;pET21a-TY 1μL;10xQ5聚合酶缓冲液5μL;Q5聚合酶1μL。设置程序:98℃预变性2 min;98~C变性15 s,66℃退火15 s,72℃延伸2 min,30个循环;72℃再延伸2 min;16℃保存,完成后取3tμL琼脂糖凝胶电泳验证扩增结果。
扩增产物经过琼脂糖凝胶回收纯化后,与抽提好的pPIC3.5K质粒同时进行EcoRI与NotI双酶切,酶切体系:ddH20 23 ILL;TT或pPIC3.5K 20μL;EcoRI IμL;NotI1μL;10xCutsmart 5μL。设置酶切程序:37℃酶切1 h,80℃灭活15 min。酶切产物经由核酸吸附柱回收后,测定浓度。按如下体系加入样品进行酶连;TT 8.5 μL;pPIC3.5K 8.5 μL;连接酶1μL;10x连接酶缓冲液2μL。设置酶连程序:16℃ 3 h,65℃30 min。酶连完成后全部转化200μL感受态大肠杆菌中,涂布带氨苄的LB平板,37℃培养活化12~16 h,挑取转化子以TF-EeoRI-F与3AOX为引物进行菌落PCR,琼脂糖凝胶电泳验证转化子。挑取能扩增出正确条带的转化子于带氨苄抗性的LB培养液中,放置于摇床37℃,220 rpm培养12~16 h。提取质粒。测定浓度并送去测序。
3、重组酵母菌株的构建与验证。毕赤酵母感受态制备,质粒的线性化与毕赤酵母的电转参照文献2中的方法进行。将长出的毕赤酵母转化子点到提前准备好的浓度为0.5 g/L的G418抗性平板上,做好标记。30℃培养3~5d后正常生长出的转化子继续点到0.75 g/L的G418抗性平板上,逐步提升浓度筛选高拷贝转化子。用无菌牙签将高拷贝转化子挑到5μL无菌水中混匀,放置在PCR仪中95℃处理5min。然后取其中1μL作为模板进行PCR。以5AOX与3AOX为引物,验证基因是否插入基因组。
4、重组酵母菌株的诱导表达。将高拷贝转化子及野生型GS115接种至50 mL(500 mL锥形瓶)BMGY培养液中,放置于摇床,30℃下250 rpm培养至0D600=2~6(16~18h),12000 rpm室温离心5 min收集菌体。用无菌水12000 rpm室温离心5 min清洗三次去除残余甘油,最后用100 mL BMMY重悬细胞。于30℃,250 rpm的条件下诱导表达,每24 h取500μL菌液并补加500 μL甲醇。将取得的菌液12000 rpm离心2min,用40 txL无菌水重悬,加入10μL 5×蛋白上样缓冲液后煮沸10 min,12000 rpm离心10 min,取10μL样品进行聚丙烯酰胺凝胶电泳,分析目的蛋白的表达水平。
二、结果与分析
(一)目的基因的验证
如图2-1所示,对空载的质粒pET21a和含有普鲁兰酶(pulA)的重组质粒pET21a-TT进行琼脂糖凝胶电泳检测,可以看到pET21a-TT因为质量分数较大所以其条带迁移距离相对于空载质粒更短,且提取的质粒线性条带位置符合理论值,用软件DNAman对测序结果的序列进行比对,比对结果发现,测序结果显示序列是完全正确的。说明含有目的基因的重组质粒经过验证可以继续进行实验。
(二)重组质粒的构建
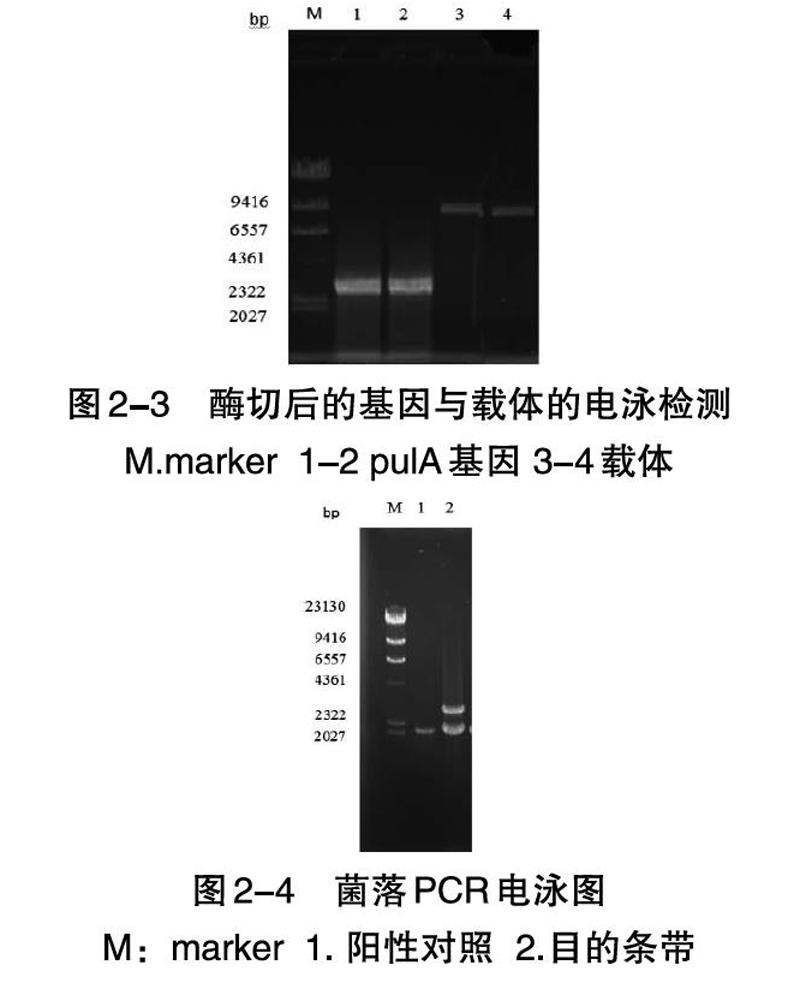
如图2-2所示,基因的条带迁移距离相对于Marker条带的位置符合其序列长度,目的条带切胶回收后测序结果正确。证明已经成功的扩增出含有双酶切位点的线性普鲁兰酶基因。如图2-3所示,基因和载体双酶切之后进行琼脂糖凝胶电泳檢测,目的pulA基因大小是2556 bp,载体大小是9004 bp,对照marker条带的结果,基因条带位置与预期结果基本相同,证明基因和载体在大小上基本正确,可以进行下一步酶连。
(三)重组酵母菌的构建与验证
将含有正确重组质粒的大肠杆菌进行大量的质粒抽提,使用SacI线性化后电转入毕赤酵母,涂布MD平板,30~C放置2~3d长出转化子。对其中的高拷贝阳性转化子进行了筛选,挑取能在2 g/L浓度的G418 YPD平板上生长的单菌落进行菌落PCR。菌落PCR电泳结果如图2-4所示,可以看出扩增出了目的基因条带,而且在2.1 kb处有明显的甲醇利用快速型(Mut+)的特异性条带醇氧化酶1的基因(aoxl)。
(四)诱导表达结果的分析
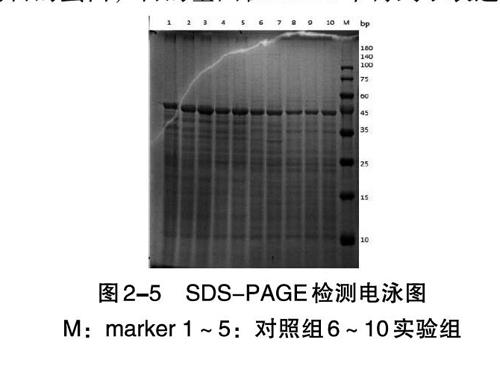
如图2-5所示,在甲醇诱导过的SDS-PAGE电泳图中可以观察到诱导时间为48 h的时候隐约出现一条微弱的条带,条带所处的位置对应marker的条带位置符合目的蛋白分子量,大小是109kDa。说明在该位置出现的可能是本实验中的目的蛋白,目的基因在GS115中得到了表达。从左至右1~5,6~10依次为每隔24h取得的样品。
三、结论与讨论
本实验验证了基因和载体序列的正确性,成功构建了重组质粒,对其进行线性化后转化毕赤酵母,获得的转化子进行鉴定并测序,确认该基因序列正确,成功筛选到阳性转化子。对毕赤酵母进行普鲁兰酶的诱导表达,采用考马斯亮蓝法对不同时期的菌液蛋白质含量进行SDS-PAGE分析,实验确定目的蛋白的大小理论值在109 kDa左右。
通过SDS-PAGE分析的结果来看,曾尝试加大点样量反复进行实验,结果仍显示条带微弱,蛋白质表达量不高,分析可能是由于Pichia pastoris存在的密码子偏好性。不同种生物对于密码子的偏好存在区别。我们的目的基因与Pichia pastoris对于密码子偏爱性存在着一定的差异。对于Pichia pastoris而言,有25个是其偏爱的密码子。若本实验所需要的目的蛋白用到Pichiapastoris中的大量稀有密码子,可能会对翻译过程的顺利进行产生影响,导致相应的tRNA不足以合成本实验中的目的蛋白。可尝试解决的方案为:对密码子进行优化,将基因进行重新设计,过程中尽量使氨基酸组成与原来的氨基酸组成相同,通过把编码外源基因的密码子优化为Piehiapastoris偏爱的密码子使蛋白表达量增加;选用pPIC9K作为表达载体,尝试将目的基因进行胞外表达。
猜你喜欢
小天使·二年级语数英综合(2020年5期)2020-12-23
电脑报(2020年28期)2020-07-31
学苑创造·A版(2016年10期)2016-11-19
计算机应用(2016年7期)2016-07-19
发明与创新·中学生(2016年3期)2016-03-29
小资CHIC!ELEGANCE(2015年14期)2015-09-23
小资CHIC!ELEGANCE(2015年15期)2015-09-01
时代英语·高二(2015年2期)2015-05-18
哈尔滨理工大学学报(2014年3期)2015-01-04
环球时报(2009-07-21)2009-07-21
