
大黄素红外光谱的理论研究
2020-08-24 12:56梁小蕊方啸天牛妍懿
当代化工 2020年6期
梁小蕊 方啸天 牛妍懿
摘 要:为了更好地了解大黄素的物理化学性质,在Gaussian09程序包中,采用密度泛函理论(DFT),B3LYP/6-31G方法对大黄素的基态最稳定结构进行了优化,研究探讨了大黄素的结构特点。并在其稳定构型的基础上,用B3LYP/6-31G方法计算了大黄素分子的红外振动光谱,根据振动强度的不同将其红外光谱分成三个区域,分析了各区域的振动情况。
关 键 词:大黄素;密度泛函理论;红外光谱;振动频率
中图分类号:O657.3 文献标识码: A 文章编号: 1671-0460(2020)06-1131-04
Theoretical Study on the Infrared Spectra of Emodin Molecule
LIANG Xiao-rui1, FANG Xiao-tian2, NIU Yan-yi1
(1. Naval Aviation University, Yantai Shandong 264001, China;
2. Cadet Team 26, Naval Aviation University, Yantai Shandong 264001, China)
Abstract: Emodin is a kind of hydroanthraquinones compound with extensive pharmacological action. In order to better explore the physical and chemical properties of the emodin, the most stable configuration of the emodin was optimized with the method of density functional theory (DFT) at B3LYP/6-31G level. The characteristics of the optimized configuration of the emodin were studied. Based on the configuration of optimization, the infrared spectra (IR) of emodin molecule were calculated by DFT B3LYP/6-31G method. Then the data of IR were analyzed, and the peak of the spectrum was relegated.
Key words: Emodin; Density functional theory; Infrared spectra; Vibration frequency
大黃素( emodin)是一种来源广泛的羟基蒽醌类化合物, 其化学名称是1,3,8-三羟基-6-甲基蒽醌,分子式是C15H10O5,状态为针状结晶,在丙酮中呈橙黄色结晶,在甲醇中呈黄色结晶。是中药大黄的主要有效成分之一,在决明子、首乌藤等多种中药材中也有丰富的含量 [1-4],在某些海洋真菌次生代谢产物中也有所见[5-6]。大黄素是一种酪氨酸激酶抑制剂,其药理作用广泛, 可用作泻药,还具有抑菌、止咳、利尿、抗肿瘤、降血压等作用[7-9]。国内外关于大黄素的研究不断深入,报道也日益增多,近年来的研究发现,大黄素对肺癌、乳腺癌等多种肿瘤均有抑制作用[7]。不仅在医疗方面发挥了巨大作用,大黄素也可用于保健和日常化工品当中,例如护肤品、洗发用品中。
本文采用密度泛函理论方法,在6-31G基组水平上对大黄素进行了结构优化,基于其优化构型,计算了大黄素的振动频率,绘制了红外光谱,并对光谱数据进行了分析讨论及谱峰的归属,以期更好了解大黄素的各类物理和化学性质,为大黄素的应用提供一定的理论指导。
1 计算部分
用Gaussian View软件构建了一种羟基蒽醌类衍生物大黄素分子的初始构型。然后用密度泛函理论,杂化密度泛函B3LYP方法,对大黄素分子的初始稳定构型进行了结构优化[10]。在优化结构的基础上,仍采用B3LYP方法,选取6-31G基组,计算了大黄素分子的红外振动频率,绘制了红外谱图,并进一步探讨了大黄素红外光谱振动频率的归属。全部计算采用Gaussian09程序包完成。
2 结果与讨论
2.1 分子的几何构型
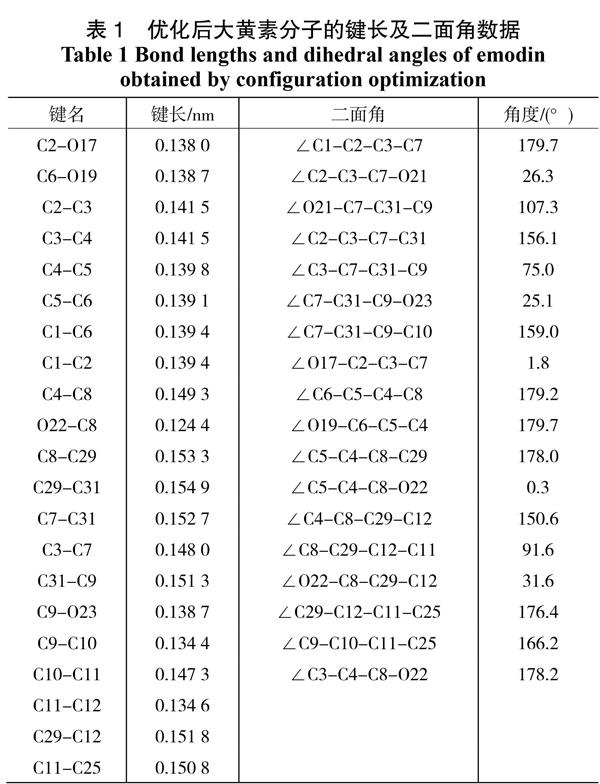
采用密度泛函理论方法对大黄素基态分子进行了几何结构优化。图1给出了大黄素分子的平面结构,图2给出了优化后的大黄素分子立体构型,及其笛卡尔坐标系。由图可见分子中的1,4-环己二酮环与苯环基本在同一平面,而与甲基取代的六元环明显不在一个平面。表1给出了优化后分子的部分结构特征参数。
从表1的键长数据来看,优化后苯环中的碳碳键长为C2–C3 =0.141 5 nm,C3–C4 = 0.141 5 nm,C4-C5 = 0.139 8 nm,均比一般的苯环键长0.139 5 nm要长,而C5-C6 = 0.139 1 nm,C1-C6 = 0.139 4 nm,C1-C2 = 0.139 4 nm,这些键长比未取代的苯环键长略短,由此可见,取代基的存在使得苯环形状扭曲,并且由上述数据可以看出两个羟基的取代对苯环形状的影响较小,而1,4-环己二酮对苯环结构影响较大;未取代时1,4-环己二酮中碳碳单键键长为0.154 0 nm,而大黄素中C3-C7、C4-C8、C7-C31和C8-C29的键长分别为0.148 0、0.149 3、0.152 7,和0.153 3 nm,可见在苯环和六元环的影响下,1,4-环己二酮也出现了扭曲,并且苯环对其影响更大。分析二面角数据可知∠C2-C3-C7-O21和∠C2-C3-C7-C31分别为26.3°和156.1°,而∠C5-C4-C8-O29和∠C5-C4-C8-O22为178.0°和0.3°,这说明苯环与1,4-环己二酮环并不完全在同一平面上,二者之间在C2-C3和C7-C31之间存在着26.3°的夹角;二面角∠C3-C7-C31-C9 = 75.0°、∠C7-C31-C9-O23 = 25.1°、∠C8-C29-C12- C11 = 91.6°、∠O22-C8-C29-C12 = 31.6°,從这些数据可以看出1,4-环己二酮环与右边六元环扭曲角度非常大。
2.2 振动频率分析
经理论优化后分子的振动频率是否存在虚频,是判断分子结构是否稳定的一个重要依据,若无虚频则说明理论优化的构型是稳定的。本文计算的大黄素优化构型的5个最小振动频率值为:29.65,50.57,70.40,91.86和127.61,它们对应的振动强度分别为:3.315 8,0.556 7,0.775 7,1.182 7和2.370 7。计算结果中没有出现虚频,说明本文所采用的优化方法合理,所得构型是势能面上的稳定点。
2.3 前线分子轨道分析
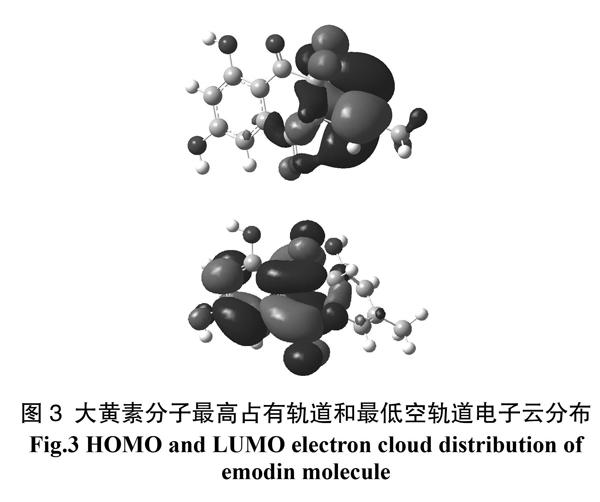
通过上述计算得到大黄素分子优化后的能量为-25 983.03 eV,能量很低,说明优化后的分子构型已达到其最低能量状态。利用Gaussian view程序绘制了分子的最高占有轨道(Highest Occupied Molecular Orbital, HOMO)和最低空轨道(Lowest Occupied Molecular Orbital, LUMO)的电子云分布图(图3),由图3可知该分子HOMO电子云主要分布在分子右侧的六元环上,而LUMO电子云主要分布在分子左侧苯环和中间六元环上,因此整个分子形成了两个π共轭体系。
同时得到的HOMO能量E = -8.59 eV, LUMO能量E = -5.96 eV,二者差值为2.63 eV,能级差较小,说明大黄素化合物中存在离域大π键,使得能级较小,离域π电子容易被激发。
2.4 分子红外光谱分析
利用密度泛函理论,B3LYP方法计算出的大黄素分子的红外振动光谱见图4。
按照吸收峰的情况,将谱图分成三个区域:300~1 000、1 000~1 710、3 000~3 700 cm-1分别讨论,由于0~300、1 710~3 000 cm-1这两个区域内几乎无吸收峰,不做讨论。
300~1 000 cm-1区域内的最强峰出现在414 cm-1处,主要是由苯环上的两个羟基中18H和20H的面外弯曲振动引起的,图5给出了它们的振动模式;次强峰出现在438 cm-1处,是由右边六元环上的羟基氢24H的面外弯曲振动产生的,如图6所示; 849 cm-1处的中等强度的峰是由苯环上的13H和14H引起的,如图7;880 cm-1处的中强峰主要是8C、4C、14H和32H的面内弯曲振动引起的。
1 000~1 710 cm-1范围内的红外吸收峰数目是三个区域内最多的,并且峰强度也是三个区域内最强的。这一区域内的最强峰出现在1 617 cm-1处,它也是整个大黄素红外光谱中的最强峰,是由2C-3C、5C-6C的伸缩振动和13H、18H、14H、20H的面内弯曲振动引起的,见图8;次强峰在1 171 cm-1处,是13H、20H和24H的面内弯曲振动引起的;1 297 cm-1处的较强峰是由13H、16H、15H、18H、24H、30H、32H的弯曲振动引起的;在1 632~1 710 cm-1范围内的几个较强吸收峰都是由9C=10C、11C= 12C、7C=21O、8C=22O等双键的伸缩振动引起的。
3 000~3 700 cm-1区域内的吸收峰是最少的,强度也较小,主要振动类型为伸缩振动。其中3 681、3 686和3 699 cm-1处的较强吸收峰分别为羟基23O-24H、17O-18H和19O-20H的伸缩振动引起的,如图9、图10、图11所示。
3 结 论
本文选取了一种药理活性非常广泛的羟基蒽醌类化合物——大黄素进行分子结构及光谱的理论研究。全部量子化学理论计算在Gaussian09软件中进行,利用DFT中的B3LYP方法,以6-31G为基组,优化得到了大黄素分子的最稳定几何构型,并得到了分子总能量和最高占有轨道、最低空轨道的能量差;用Gaussian view程序绘制了大黄素分子的最高占有轨道和最低空轨道的电子云分布图,结果显示用以上方法得到的分子结构是稳定的。在优化稳定构型的基础上,用同样的方法计算了大黄素分子的红外振动频率,并用Gaussian view程序绘制了红外光谱图,根据红外吸收峰强度的不同,将大黄素的红外光谱分成三个区域,分别探讨了它们的振动模式。结果表明1 000~1 710 cm-1范围内的红外吸收峰数目和强度是三个区域内最多、最强的。最强峰出现在1 617 cm-1处,是由2C-3C、5C-6C的伸缩振动和13H、18H、14H、20H的面内弯曲振动引起的;300~1 000 cm-1区域内的吸收峰较少,最强峰出现在414 cm-1处,由苯环上的两个羟基中的18H和20H的面外弯曲振动引起的; 3 000~3 700 cm-1区域内的吸收峰是三个区域中最少的,强度也较小,主要振动类型为伸缩振动。
参考文献:
[1]梁英,喻大昭. 大黄素的化学合成及结构修饰研究进展[J]. 有机化学, 2011, 31(8): 1324–1333.
[2]刘静,王丽. 大黄素的研究进展[J]. 中国药房,2014,25(35):3351–3354.
[3]孙桂斌,张萌,严方,等. 大黄素抗肿瘤活性及相关机制研究进展[J]. 药学进展,2013,37(6): 248–256.
[4]高红刚,周菊华. 大黄素抗炎作用及相关机制研究进展[J]. 济宁医学院学报,2016, 39(5): 348–352.
[5]KHAMTHONG N, RUKACHAISIRIKUL V, TADPETCH K ,et al. Tetrahydroanth-raquinone and xanthone derivatives from the marine-derived fungus Trichoderma aureoviride PSU-F95[J]. Archives of Pharmacal Research, 2012, 35(3): 461-468.
[6]安昶亮,孔凡栋,马青云,等. 海洋真菌Aspergillus sp. SCS-KFD66 的次级代谢产物研究[J].中草药,2019,50(13): 3001–3007.
[7]林玩福,汪晨,凌昌全. 大黄素抗肿瘤作用研究进展[J]. 中国中药杂志,2015,40(20): 3937–3940.
[8]刘瑞霞,齐文杰. 大黄素治疗重症急性胰腺炎的作用与机制研究进展[J]. 临床和实验医学杂志,2016,15(2): 193–195.
[9]SONG H Y,ZHANG L,PAN J L,et al. Bioactivity of five components of Chinese herbal formula Jiangzhi granules against hepatocellular steatosis[J]. Journal of Integrative Medicine,2013,11( 4) : 262.
[10]梁小蕊,孫晓伟,刘存海,等. 一种海洋天然产物分子的红外光谱及核磁共振碳谱的理论研究[J].当代化工,2018,47(12):2542-2544.
猜你喜欢
中学课程辅导·高考版(2020年9期)2020-10-20
科技视界(2020年19期)2020-07-30
中学课程辅导·教学研究(2017年29期)2018-02-26
中学化学(2017年3期)2017-03-28
视野(2016年6期)2016-05-14
食品界(2016年6期)2016-05-14
读者(2016年4期)2016-01-26
意林·少年版(2014年9期)2014-07-28
数理化学习·高一二版(2009年1期)2009-03-19
中国科技术语(2004年4期)2004-03-18
