
TREM2在阿尔兹海默症中的研究进展
2020-08-07 07:11李晓月倪赛佳姚增莹张启春
中国药理学通报 2020年8期
李晓月,倪赛佳,姚增莹,张启春
(南京中医药大学药学院,江苏 南京 210023)
阿尔兹海默症(Alzheimer′s disease,AD)是一种隐匿发生且发展缓慢的神经退行性疾病,最初在一个多世纪前由德国神经精神病学家Dr.Alois Alzheimer发现,到如今已被认为是痴呆性疾病中最常见的类型。其主要两个病理特征为胞外淀粉样-β(amyloid-β,Aβ)斑块沉积及异常tau蛋白高磷酸化所致的神经纤维缠结。此外,AD还伴有脑内神经炎症的发生,主要由脑内定居小胶质细胞对病理产物的免疫应答所致。根据临床表征及遗传风险程度,通常将AD分为3种类型:家族性AD、早发型AD及迟发型AD。已经明确的是家族性AD由编码淀粉样前体蛋白(amyloid precursor protein,APP)、早老素1(PSEN1)、早老素2(PSEN2)的相关基因突变所引发。然而对于通常发生在65岁之后的迟发型AD,虽在AD患者中占比很大,却并不表现携带任何家族性AD相关的基因突变,其发病原因目前尚不完全清楚,一般认为是多种因素(包括生物因素和社会因素)综合作用下的结果。近年来发现TREM2编码区的罕见突变可显著提升阿尔茨海默病的患病风险[1],因此对于TREM2的功能及其调节下的小胶质细胞在中枢神经系统疾病发生发展中的作用日渐成为关注的焦点。
1 TREM2的结构、配体及其相关信号通路
1.1 TREM2结构TREM2属于免疫球蛋白超家族的一个单程传递膜受体,最初在单核细胞来源的树突状细胞和小鼠巨噬细胞中发现。TREM2基因位于人类6p21.1染色体,小鼠17C3染色体上具有免疫功能的基因簇内,其在外周主要于髓系细胞表达,如巨噬细胞、树突状细胞、粒细胞等。而在中枢神经系统,TREM2只由发挥主要免疫作用的小胶质细胞表达,且转录信号强度在基底神经节、胼胝体、延髓和脊髓显示最强[2]。TREM2结构包括3部分:胞外区、无信号基序的跨膜结构域和胞质内部分,此外其跨膜区域还通过带正电的赖氨酸残基与DNAX活化蛋白12(DAP12,也称作TYROBP)及DNAX活化蛋白10(DAP10)相关联。TREM2胞外区包括单个免疫球蛋白结构域和柄区,研究发现在髓样细胞受到诸如Toll样受体配体或炎性细胞因子刺激信号激活后,胞外结构域会受到α-分泌酶解体蛋白ADAM17或ADAM10在柄区处的切割,释放可溶形式的TREM2(sTREM2)至胞外,使细胞表面膜结合TREM2水平迅速降低[3]。
1.2sTREM2关于sTREM2发挥的生物学功能尚无确切定论,有报道表明这种TREM2经切割后的可溶形式产物是有一定活性的。研究发现,sTREM2可能会通过激活Akt-GSK3β-β-catenin信号通路减少小胶质细胞凋亡并依赖于 PI3K/Akt信号通路促进小胶质细胞的存活,发挥神经保护效应。此外,研究还表明,不论正常小鼠还是TREM2敲除小鼠,在向海马区输入sTREM2-Fc蛋白后,均能提高小鼠炎性因子表达水平,同时也会使小胶质细胞的形态发生改变,免疫应答能力增强。该文章提示增强sTREM2信号通路或许可作为AD的一种治疗方式[4],他们通过向5xFAD小鼠直接定向注射重组sTREM2蛋白或是腺病毒转染的方式提高sTREM2的水平,发现其能促进淀粉样斑块附近小胶质细胞的增值及吞噬活性,加快对斑块的吞噬清除而发挥保护作用。sTREM2能够通过脑脊液-脑屏障,可在脑脊液中检测到。当前已确证sTREM2可用来当作AD疾病检测的标志物[5],根据2018年发表的一篇报道,脑脊液sTREM2浓度与神经变性程度及tau蛋白纤维病理学指标相关,表明脑脊液sTREM2的水平可作为AD的临床分类标准。而在最近的一篇文章,作者对携带6种不同TREM2罕见突变的AD患者的sTREM2的水平研究,发现各类TREM2突变下sTREM2表达水平异于TREM2突变不携带的AD患者,其中R47H突变的患者CSF中sTREM2平均水平明显较其他类型突变者高。而至于生成增加的sTREM2是否使R47H突变患者有别于TREM2正常的AD患者还有待探究。此外,研究者在将病人根据AD不同临床阶段分类后,分别检测了sTREM2与Aβ病理相关标志物及Tau蛋白病理相关标志物的关联。结果表明,AD中sTREM2水平响应于Tau蛋白病理程度加重升高,而似乎不受Aβ病理状况的影响[6]。对于sTREM2与Tau蛋白之间的联系亦仍需探究。此外sTREM2在正常状态的脑组织中也发挥着不可忽视的作用,在针对sTREM2与大脑结构关系的研究中,研究发现,CSF中sTREM2的表达水平能够改变认知未受损的健康成年人脑容量及脑组织迁移率。
1.3 TREM2相关配体TREM2带正电的功能部位会基于电荷基础与其阴离子配体结合而被激活,其常见的配体有:细菌脂多糖、磷脂、载脂蛋白、细胞碎片、Aβ蛋白等。TREM2能够与多种革兰氏阴性菌、革兰氏阳性菌及酵母菌结合,并促进细胞对其吞噬。TREM2敲除后,细胞对细菌的吞噬率会显著降低,而在将TREM2和DAP12嵌合体导入非吞噬细胞后则可促进其对细菌的吞噬作用。TREM2与磷脂的结合也被广泛证实,在小鼠的AD模型中发现,在Aβ蛋白积累及神经元丢失的诱导下会导致某些脂质的释放,包括来自受损的神经元或胶质细胞的膜磷脂,如磷脂酰丝氨酸,这些脂质与TREM2结合后会激活TREM2受体,导致小胶质细胞的增殖效应的发生[7]。载脂蛋白E(APOE)是近年来发现的TREM2新型配体,其等位基因APOE4突变是研究最为明确的迟发型AD致病风险因子。去年Wang等[8]首次在人类系统模型上证实了APOE4导致AD的机制,他们利用源于诱导多能干细胞的表达ApoE4的人类神经元建立细胞模型,发现 ApoE4突变下神经元会有更严重的Tau蛋白磷酸化及Aβ蛋白水平,能够导致GABA能神经元的退化,且此两种病理下的产物不显示相关性[8]。脑组织中,APOE由胶质细胞,且主要是由星形胶质细胞分泌,并经三磷酸腺苷结合盒转运蛋白A1(ABCA1)及三磷酸腺苷结合盒转运蛋白G1(ABCG1)脂质化[9]。生化检验分析发现,APOE与人类TREM2的结合具有高亲和力[10],此种结合可增加小胶质细胞对APOE结合的凋亡神经元的吞噬作用,而在TREM2突变下结合力会显著降低[11]。
1.4 TREM2的相关信号通路DAP12是多种偶联跨膜受体(如:CSF1R、TREM2)的接头蛋白,也是目前发现的TREM2唯一的接头蛋白,因此其在TREM2向胞内传递信息的过程中起到至关重要的作用。DAP12的突变也会直接导致某些疾病的发生,如:NHD[12]。在TREM2-DAP12-DAP10信号通路中,DAP12上所含有的免疫受体酪氨酸激活基序(ITAM)磷酸化之后会通过募集酪氨酸蛋白激酶(SYK)向胞内传递信号,进而激活下游多个通路,如:细胞外调节蛋白激酶(ERK)、胞内磷脂酰肌醇激酶(PI3K)、丝氨酸/苏氨酸激酶(Akt),从而可能促进细胞表面TREM2的表达,促使小胶质细胞向吞噬及抗炎表型转化[13]。而DAP10则在TREM2-DAP12信号通路募集PI3K的过程中发挥关键作用。有研究表明,含有Src同源结构域2(SH2)的物质,诸如肌醇磷酸酶-1(SHIP1),则可以通过Src同源结构域2(SH2)与DAP12结合抑制PI3K的募集。而在γ-分泌酶作用下TREM2膜结合区域会发生降解,进而导致TREM2信号的减弱[14](Fig 1)。有研究者认为对α-分泌酶及γ-分泌酶的抑制也是AD的一种潜在治疗手段[15]。
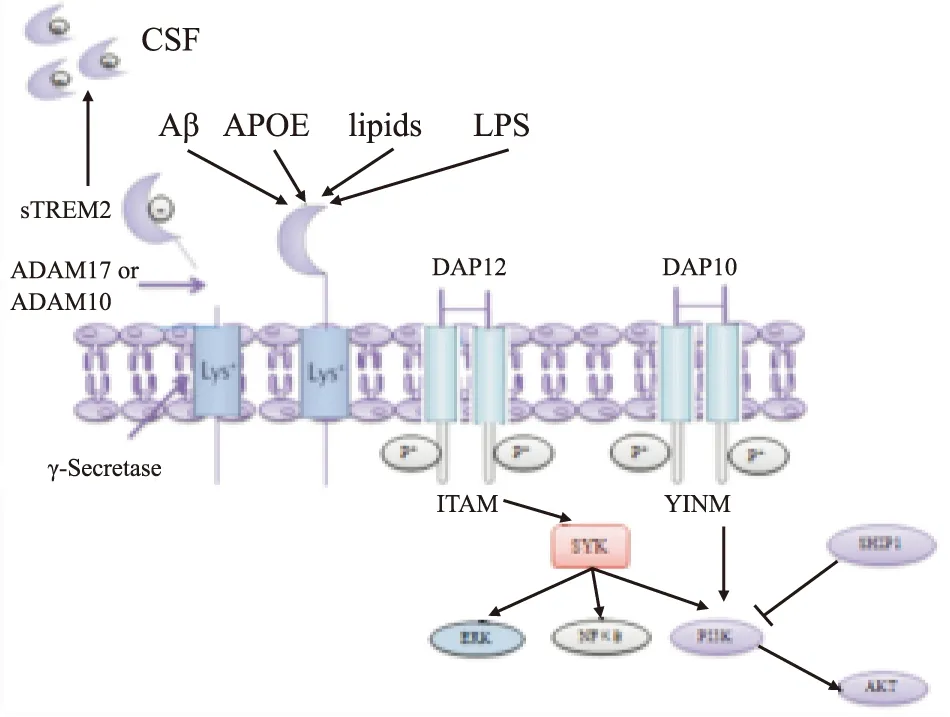
Fig 1 Structure and signaling pathways of TREM2
2 TREM2对小胶质细胞功能的影响
小胶质细胞是中枢神经系统的定居免疫细胞,正常状态下约占中枢神经系统总细胞的10%,主要分布在黑质区域。它们最初起源于卵黄囊祖细胞,这些祖细胞会在胚胎发育第8.5 d通过血液循环迁移到发育中的中枢神经系统,且此过程会一直持续到中枢神经系统形成之前。不同于中枢神经系统中的其它细胞(包括大胶质细胞、神经元)均由神经外胚层分化而来,小胶质细胞来源于外周中胚层[17],它们具有终生的分裂能力,无需来自外周髓系细胞的补充,虽在数量上并不显优势,但却是中枢神经系统内的主要免疫细胞。
过去很长时间里,小胶质细胞所发挥的作用被严重低估。近年来随着小胶质细胞更多的功能被逐步发现,表明其除了像巨噬细胞一样发挥免疫作用外,还会通过直接接触与神经元细胞交流,参与神经元存活、凋亡、迁移、突触修剪、甚至神经回路形成等的生理活动[17-19]。正常状态下小胶质细胞呈分支状,会利用胞体伸出的长分支不断运动来监测脑环境,接受胞外信号并对其进行转导整合,不断对自身调整以维持脑内稳态,此时也会有一小部分小胶质细胞在有限的时间里显示吞噬活性来将凋亡的细胞清除。而在受到损伤或感染的情况下,小胶质细胞则会应对损伤分子相关模式(PAMPs)或病原相关分子模式(DAMPs)做出应答,在病灶局部逐步激活向疾病相关小胶质细胞表型转化,形态上有胞体变大、分支减少且加粗的变化,表现为一种过度增大的或是阿米巴样的小胶质细胞。此过程取决于特定的病理状况,不同病理环境下转化而成的不同小胶质细胞表型发挥的效应可能有较大差异。此时小胶质细胞还会发生活跃的增值,形成小胶质细胞屏障减缓病灶区的扩大,同时吞噬能力也会增强,发挥一定的神经保护作用[20]。
TREM2作为一个具有免疫调节作用,在CNS中只表达于小胶质细胞的受体,能够通过参与小胶质细胞的吞噬、增值、存活、自噬、能量代谢等方面而发挥作用。在TREM2纯合子T66M和W50C错义突变引起发病的NHD患者中获取并诱导产生多能干细胞来源的小胶质样细胞,发现携带TREM2突变的小胶质样细胞的TREM2表达和分泌水平显著降低,且细胞存活率下降,对凋亡体的吞噬作用受损,显示TREM2在维持小胶质细胞存活及正常吞噬方面的重要作用[21]。研究发现,在TREM2敲除的5XFAD小鼠模型中,小胶质细胞会产生大量的自噬小泡,在加入环肌酸增加能量储存后有所减轻,表明在TREM2敲除小鼠中小胶质细胞有能量代谢障碍的发生[22]。也有报道称,TREM2可能与细胞凋亡也有关联,在对铜绿假单胞菌所致角膜炎的研究中,研究者发现Trem2-/-小鼠会较正常C57BL/6小鼠病情加重,最终证实此效应是由于TREM2能够通过抑制caspase-1依赖的细胞凋亡来促进宿主对铜绿假单胞菌的抵抗而产生的[23]。另外,大量研究表明,在TREM2敲除的状态下,会损伤小胶质细胞在病灶区域附近的增殖激活,从而在数量及效应上使小胶质细胞不能发挥正常的脑保护作用,加重疾病损伤。
3 TREM2与神经系统疾病
研究发现TREM2在诸如阿尔兹海默症(AD)、帕金森症(PD)、自闭症(ASDs)、多发性硬化(MS)、肌萎缩侧索硬化症(ALS)等众多神经系统疾病中均会表达上调[24]。
3.1AD目前已大约发现30个与AD发病相关的常见基因突变位点,其中载脂蛋白E基因的ε4等位基因突变是目前研究最多最明确的迟发性AD风险因子。此外也发现一些编码免疫调节作用蛋白基因(譬如,TREM2、CD33)的罕见突变也会增加迟发型AD的发病风险,关于TREM2与AD的关联研究目前备受关注。运用基因组测序、外显子组测序及桑格测序法,对1 092名AD患者及1107位正常受试者TREM2遗传变异性进行分析,发现AD患者中共有22名TREM2等位基因突变者,而对照组中仅有5名,显示 TREM2突变与AD发病存在着高度相关性。之后的全基因组关联研究也证实了TREM2的罕见突变会显著增加迟发型AD的发病风险[25]。另外,在发表于2017年的一篇报道中,用单细胞RNA测序的方法在AD模型中发现了一种具神经保护作用的疾病相关小胶质细胞新表型。他们发现这类新发现的疾病相关小胶质细胞激活过程包括2个阶段,在激活的第1阶段虽不依赖TREM2,但会增加TREM2信号结合配体相关基因的表达,例如:APOE。而其激活的第2阶段则依赖于TREM2信号通路,此阶段的细胞中与脂质代谢及吞噬相关的信号通路会表达上调,在TREM2敲除的情况下第2阶段激活过程则会发生障碍,暗示了 TREM2对于AD疾病发展晚期阶段可能具有重要意义。最为重要的是,这篇报道发现的与AD相关的小胶质细胞新表型所具有的对神经退行性疾病的抑制作用[26],也显示了这种疾病相关小胶质细胞作为一种治疗迟发型AD手段的可能。
那么TREM2究竟在AD中扮演怎样的角色呢?TREM2能作为小胶质细胞上Aβ斑块的受体,在与Aβ结合后负责下游信号的传导,TREM2完全敲除会导致Aβ诱导分泌的细胞因子及下游信号通路改变,进而使小胶质细胞对病理产物的吞噬作用受损。研究工作也证实,TREM2在清除病理产物上的重要贡献,他们发现TREM2缺失下会降低运用Aβ抗体结合促进小胶质细胞对Aβ斑块清除的AD免疫疗法疗效。在TREM2敲除下会加重5XFAD小鼠模型的Aβ斑块沉积及神经元丢失状况。而就针对TREM2部分缺失及完全缺失的差异,通过RNA测序发现TREM2单倍剂量不足与完全缺乏状态下小胶质细胞转录组差异极大,于是随后观察了在体情况下Tau蛋白病理模型P301S+小鼠TREM2部分缺失和完全缺失小胶质细胞应对病理损伤的差异,发现对疾病应答的过程中TREM2+/-小鼠比Trem2-/-小鼠的小胶质细胞显示更强的损伤作用,会加重Tau蛋白病理状况,且这种效应呈现年龄依赖的趋势,在老龄鼠中表现显著。而相比之下,TREM2完全缺乏却显示一定的脑保护作用,此组别的小鼠在应对Tau蛋白所致的脑萎缩下侧脑室体积会有所减小,研究者猜测这种效应是由于TREM2完全缺失后小胶质细胞增值、激活。或许TREM2突变就好比一个司令部总在发出错误指令一样,会导致比完全瘫痪更为可怕的后果。而由于杂合子突变似乎显示在迟发型AD发病缘由中占有很大比例,因此单倍剂量不足所致的效应可能更有参考意义。
关于TREM2单个核苷酸多态性(SNPs)与迟发型AD的关联的研究,以R47H突变最为受关注,其增加AD发病风险的让步比为5.05,几乎与APOE4风险因子相当。早前有研究者在体外试验中发现R47H突变会损伤TREM2与配体的正常结合功能,近年来陆续有报道阐明在体状态下R47H对AD的作用。在APPPS1-21,R47H突变小鼠中,总TREM2 mRNA的表达水平有明显下降,斑块附近的小胶质细胞对沉积的淀粉样蛋白反应性降低,斑块周围小胶质细胞积累减少,且与TREM2敲除组小鼠相比无显著差异[27]。而在另一篇运用人源化淀粉样蛋白病理模型的报道中也观察到同样的效应,作者对比了5XFAD;R47H小鼠、5XFAD;Trem2-/-小鼠和普通突变的5XFAD小鼠在对于淀粉样斑块积累过程中的应答差异,发现 R47H突变会损坏病理性斑块诱导下所致的小胶质细胞在斑块附近的增殖激活,效应与Trem2-/-;5XFAD组别亦无显著差别,而普通突变组小胶质细胞在斑块附近的增殖效应却并没有受到影响[28]。总的来说,从研究结果可以推测R47H突变增加AD患病风险的缘由可能是R47H突变下会显著降低TREM2与相关配体结合,从而可能损害到小胶质细胞的正常功能,使其不能及时对某些风险信号做出应答以维持脑内稳态。而当疾病开始发生时,小胶质细胞也无力应答病理斑块做出正常应答来减缓疾病进程的发展。
3.2 其他相关疾病虽研究的聚焦点主要落在TREM2单个核苷酸多态性与AD间的关联上,TREM2突变与其他一些神经退行性疾病(诸如Nasu-Hakola、额颞叶痴呆、ALS)的联系也不容忽视。Nasu-Hakola(NHD)也称脂膜样多囊性骨发育不良,是一种罕见的常染色体隐性遗传病,该疾病的主要临床表现有发展迅速的痴呆症状及仅限于手腕和脚踝的骨囊肿症状。遗传学研究表明这一疾病与TREM2及其接头蛋白DAP12的失活突变相关。有研究者发现TREM2缺失下患者的破骨细胞分化会受到抑制,从而导致骨吸收活性下降,表明这可能就是NHD患者会发生反复骨折的原因。ALS也与TREM2的突变显示一定关联,从美国ALS诊所中招募到923名ALS受试者并以1 854名非西班牙裔白人作正常对照。他们对样本的TREM2的R47H变异体进行基因分型。结果发现与对照组相比,携带R47H突变的患者在ALS患者中更为普遍,让步比为2.4,表明R47H同时也为ALS的一个显著风险因子。另外,也有研究者表明TREM2突变也对自然衰老下小胶质细胞的改变及神经元丢失的发生有一定贡献[29]。
4 TREM2的变异体
TREM2突变虽然罕见,但发现的类型很多。目前与AD相关的TREM2突变已发现有46种,其中罕见变异体R47H(rs75932628),编码蛋白链上第47位精氨酸变为组氨酸的突变,被认为与迟发型AD发病高度相关。在对欧洲和北美人群的调查中发现R47H能够增加AD的发病风险到两到三倍。一般认为,R47H突变下损伤TREM2与其阴离子配体的正常结合是导致某些疾病发生的分子学机制。通过对R47H及正常TREM2对照组胞外结构域的晶体结构研究发现Arg47对于维持TREM2互补决定区2(CDR2)环的结构特征及稳定其与配体相互作用的构象上发挥重要作用。这或许也解释了为何在众多TREM2突变体中,R47H突变显示更显著后果的原因。而对于其他AD相关TREM2的变异体,虽研究相对较少,但仍显示出不可忽视的效应。研究发现位于胞外结构域上的错义突变p.T66M及p.Y38C,除明显增加TREM2蛋白的错误折叠外,还会导致未成熟的TREM2蛋白滞留于内质网以致使TREM2表达下降,影响到小胶质细胞的正常功能,包括依赖于TREM2的吞噬功能。而α-分泌酶的切割位点,157位上编码组氨酸的突变p.H157Y则会增强TREM2裂解导致TREM2表达缺失。另有报道称在对p.R62H和p.R62C两种突变体的体外研究显示,此两种突变下会对磷脂的应答下降,而另两种突变p.T96K和p.D87N虽显示会与磷脂的结合增强,但却和前两种突变产生相似的效应。此外,研究者也发现了具有神经保护作用的TREM2突变,通过全基因组关联研究对AD患者颞叶皮层(n=399)及小脑(n=374)基因表达分析,发现一种TREM2 突变(命名为rs9357347-C),可以通过提高TREM2及TREML1的表达水平来降低AD的发病风险,发挥神经保护作用。而这些及其他罕见的TREM2变异体在体内条件下的影响作用有待研究。其他与AD相关的TREM2突变有:R62H、R52H、R136W、R136Q、H157Y、W191X、E202D、H215Q、T223I[27]。对于这些TREM2低频的突变在神经系统疾病中发挥的作用,虽建立基因型-表型的关联研究较为困难,但仍具有重要的意义,此研究工作蕴藏着潜在价值可为临床上治疗疾病提供新的思路和手段。
5 总结
血脑屏障作用下各类循环中的免疫细胞难以进入脑实质,尽管研究表明激活的T淋巴细胞可以通过变形的方式穿过内皮组织进入脑内,但小胶质细胞仍旧是中枢神经系统应对不利环境下的主要免疫细胞。因此,维持小胶质细胞的健康状态应该是预防及治疗脑疾病的关键着手点。TREM2在调控小胶质细胞的生命活动方面具有重要意义,可以通过众多途径调控小胶质细胞的各类功能,其作用甚至可以跨到另一个维度上,对其他系统(如循环系统)产生某些影响。虽然有众多谜团仍待解开,但可以确定的是,TREM2异常状态下对于小胶质细胞乃至中枢神经系统具有相当不利的影响,而对于TREM2正常结构的维持也有望成为一项有效的治疗手段。
猜你喜欢
自我保健(2021年2期)2021-11-30
妇女之友(2021年9期)2021-09-26
昆明医科大学学报(2021年3期)2021-07-22
昆明医科大学学报(2020年11期)2020-12-28
神经损伤与功能重建(2020年10期)2020-12-23
神经损伤与功能重建(2020年11期)2020-12-01
无机化学学报(2020年7期)2020-07-20
物理化学学报(2020年4期)2020-04-24
渤海大学学报(自然科学版)(2020年3期)2020-02-02
百姓生活(2019年2期)2019-03-20
