
小麦属植物叶绿体基因组结构的比较分析
2020-07-31 10:41何兆峰欧平和杨玉存王鹏程崔立操聂小军
麦类作物学报 2020年1期
苏 宁,何兆峰,欧平和,杨玉存,王鹏程,崔立操,聂小军
(1.西北农林科技大学农学院,陕西杨凌 712100; 2.江西农业大学生命科学学院,江西南昌 330045)
叶绿体是植物特有的进行光合作用的细胞器,其具有自身的基因组可半自主地进行遗传物质的复制和转录,参与植物许多重要的生理生化过程[1]。被子植物的叶绿体基因组通常为环状四组分结构,包括1个大单拷贝区(large single copy,LSC)、小单拷贝区(small single copy,SSC)和2个反向重复序列区(inverted repeats,IRa/IRb),大小一般为120~180 kb[2]。与核基因组相比,叶绿体基因组小,结构稳定,遗传重组率低[3],具有很强的保守性[4],但在IR边界,不同物种又存在一定的扩张与伸缩变异,这对研究绿色植物的进化、揭示亲缘关系具有重要价值,是物种分子鉴定、系统进化和遗传多样性研究的重要信息来源[5]。
小麦是世界上最重要的粮食作物之一,其种植面积约占世界总耕作面积的17%,提供了全世界20%的粮食消耗[6]。小麦属植物作为小麦的近缘种,一直以来都是普通小麦遗传改良的重要种质资源和基因库。明确小麦属植物的系统进化关系,加强对小麦属植物的开发与利用对丰富小麦种质资源、克服小麦基因同质化和促进小麦育种均具有重要意义[7]。自2002年,小麦叶绿体全基因组序列被解析以来,前人围绕小麦及其近缘种叶绿体基因组学测序及比较分析开展了大量研究工作[8]。郭长虹等[9]对普通小麦与山羊草叶绿体基因组中的热点突变区域序列变异进行了系统分析,发现普通小麦叶绿体基因组B2片段可能起源于Ae.speltoides;杨新泉等[10]利用叶绿体SSR标记对普通小麦、斯卑尔脱小麦、密穗小麦和中国特有小麦(新疆稻麦、西藏半野生小麦和云南铁壳麦)等不同类型六倍体小麦叶绿体基因组的遗传多样性进行分析,发现斯卑尔脱小麦和西藏半野生小麦遗传多样性丰富,为不同小麦的遗传差异分析提供了依据;Bahieldin等[11]利用高通量测序技术对中国春的叶绿体基因组进行了优化,修正了IR区的长度,为利用叶绿体序列分析小麦属物种的亲缘关系提供了更准确的参考;Gornicki等[12]系统测定了小麦属和粗山羊草属的40个物种的叶绿体基因组序列,并结合叶绿体基因组学的遗传变异分析,全面研究了六倍体小麦的起源、演化和进化关系。截止目前,有关小麦属各物种间叶绿体基因组的变异及其起源与进化的研究还未见报道。本研究以已发表的14个小麦属叶绿体基因组为对象,利用比较基因组分析方法,对这些小麦属植物叶绿体基因组的基因含量、序列变异、结构特性、进化关系和RNA编辑等进行了系统的分析,以期从叶绿体角度解析小麦属物种的起源进化关系,为发掘和利用小麦属植物应用于小麦遗传改良提供依据。
1 材料与方法
1.1 数据收集
从GenBank中的细胞器基因组数据库(https://www.ncbi.nlm.nih.gov/genome/organelle/)检索并下载已发表的14个小麦属植物叶绿体全基因组序列,获取注释信息(表1)。
1.2 基因组特征与比较分析
根据各物种叶绿体基因组的注释信息,利用Excel统计14个小麦属植物叶绿体基因组的大小以及LSC、SSC和IR的长度;全基因组与编码区的GC含量;总编码以及编码蛋白质、tRNA和rRNA的基因数目;位于IR区段和含有内含子的基因数目等基本结构与组成特征信息;以中国春叶绿体基因组为参考,根据各个叶绿体基因组的注释信息,对其IR边界及其邻近基因手工绘制图谱,比较各个叶绿体基因组IR区的伸缩趋势;同时,采用基因组在线分析程序mVISTA(http://genome.lbl.gov/vista/mvista/submit.shtml)进行全基因组的多序列比对,筛选其中的变异热点区;最后,以烟草叶绿体基因组为参考,利用MAUVE工具,对14个小麦属叶绿体基因组进行多序列比对,鉴定分析其基因组的重排与倒位现象。
1.3 系统发育树的构建
以二穗短柄草叶绿体基因组(NC_011032)为外源,对叶绿体全基因组构建系统进化树。利用ClustalX 1.83软件进行多序列比对,并进行手工的校正,然后采用PAUP 4.0软件构建MP系统进化树,自展值(Bootstrap)设置为1000,通过后验概率值来表示系统发育树各分支的可信度。
1.4 RNA编辑位点的预测与比较
根据各个基因组的注释文件,提取其所有蛋白质编码基因,转化格式后提交到RNA编辑位点在线预测工具PREP RNA Editing(http://prep.unl.edu),对各物种叶绿体编码蛋白基因中的潜在编辑位点进行预测,然后比较分析小麦属各物种共同和特异的编辑位点。
2 结果与分析
2.1 基因组结构与基本特征
分析发现,14个小麦属植物的叶绿体基因组均为四分体结构,没有发现大片段缺失现象,各基因组长度介于135 897~136 157 bp之间(表1)。LSC区长度为79 998~80 256 bp,相差258 bp,其中提莫菲维的LSC区最长,达到80 256 bp。SSC区在12 790 bp左右,不同物种间仅相差几个碱基。IR区长度变异较小,为21 541~21 566 bp。由此表明,LSC区长度变异大于SSC和IR区,且基因组长度变异主要是由LSC区的变化 引起。
进一步统计各物种的编码序列长度,发现其编码序列长度存在较大的变异,其中6个物种为58 692 bp;硬粒小麦的最长,达61 746 bp,而提莫菲维的最短,只有51 441 bp。GC含量分析发现,各物种叶绿体全基因组的GC含量差异较小,均在38.3%左右,基因编码区的GC含量在 38.90%左右(表1)。基因数目分析发现,小麦属叶绿体基因组编码的总基因数介于105~115个之间,其中提莫菲维小麦包含的基因数最多,为115个。rRNA编码基因数最为保守,各物种均为4个。而蛋白质编码基因数存在一定差异,11个物种包含76个蛋白质编码基因,而西藏半野生小麦、硬粒小麦和提莫菲维小麦分别有79、80和81个蛋白编码基因。另外,7个物种的tRNA编码基因为25个,且这7个物种的基因编码数目情况完全相同,可能存在较近的系统进化关系。相关性分析发现,tRNA编码基因数与基因总数的相关性系数为0.917 29,表明小麦属叶绿体的总基因数的差异主要是由tRNA编码基因引起的。
对位于IR区的基因组成和数量进行比较分析。结果发现,小麦属叶绿体IR区的基因数目存在较明显的差异,普通小麦中国春和一粒系中乌拉尔图小麦有18个,西藏半野生小麦有19个,而二粒系小麦的硬粒小麦、科尔希小麦和提莫菲维系的提莫菲维小麦IR区的基因数目分别为23、28和24个,其余物种IR区基因数目均为16个。相较于普通小麦和一粒系小麦,二粒系小麦各物种的IR区发生了显著的扩张现象,IR区的变异可能是重要的进化事件,在小麦起源进化过程发挥了重要作用。
2.2 IR边界分析
叶绿体基因组为由LSC、SSC、IRa、IRb构成的环形结构,存在四个边界,即LSC-IRb、IRb-SSC、SSC-IRa、IRa-LSC。在植物进化过程中,叶绿体基因组IR区的扩张与收缩是植物重要的进化事件,并会引起叶绿体基因组大小和基因含量的变化。以中国春叶绿体基因组为参照,对小麦属植物叶绿体基因组IR区的扩张与收缩进行了比较分析(图1),发现小麦属植物叶绿体的4个边界(即LSC-IRb、IRb-SSC、SSC-IRa、IRa-LSC)均相对保守。14个小麦属植物叶绿体中,LSC-IRb边界与rpl22均间隔28 bp,除提莫菲维(53 bp)外,其余物种LSC-IRb边界与rps19均间隔50 bp。大部分物种IRb-SSC边界距ndhF基因有68 bp的间隔,硬粒小麦和乌拉尔图小麦与ndhF基因的间隔为67 bp,而提莫菲维小麦与ndhF基因的间隔为77 bp,且13个物种ndhF基因长度均为2 219 bp,仅提莫菲维小麦为2 216 bp。 SSC-IRa边界位于ndhH基因内,13个物种ndhH位于SSC中的部分长度为974 bp,而中国春的相对较短,只有474 bp,同时13个物种的ndhH基因位于IRa中的序列长度为206 bp,只有硬粒小麦的长度为236 bp,存在30 bp的差异,使得在IRb区产生一个ndhH假基因片段。IRa-LSC边界位于rps19基因与psbA基因之间,距离rps19的位置高度保守,14个物种均为50 bp,而距psbA存在一定差异,12个物种为91 bp,而硬粒小麦和提莫菲维小麦分别为82 bp和94 bp。
2.3 基因组序列变异分析
以中国春叶绿体基因组为参照,利用mVISTA工具进行多序列比对,并将序列相似度比对结果可视化。结果发现,14个小麦属植物叶绿体基因组序列比较保守;在psbA和psbI-psbD基因间隔区,13个其他小麦属物种与中国春相比均存在序列变异;六倍体斯卑尔脱小麦在rps3-rpl22、rpl23-ndhB存在特异的序列差异,圆锥小麦在psbI-psbD、rbcL-psaI、rpl22-rps19、rpl23-ndhB存在变异,硬粒小麦在pcbC-psbZ、rpoC2、atpI-atpH、atpF、rbcL-psaI、rpl23-ndhB、trnL存在变异,并且在106~115kb段差异非常大,科尔希小麦在rbcL-psaI存在变异,提莫菲维小麦在trnG-trnM、trnD-psbM、petN-rpoB、trnF-ndhJ、psaJ-rpl33、rpl22存在差异。从叶绿体基因组的4个组分上看,IR区序列变异最低,LSC区的变异程度最高;基因间隔区变异高于编码基因,内含子变异高于编码序列;变异度最高的是基因间隔区,如:LSC-psbA、psbI-psbD、rbcL-psaI、rpl22-rps19等,这些位点为小麦属物种的分子鉴定提供了新的位点资源。
同时,以烟草叶绿体基因组(Z00044.2)为参照,利用MAUVE软件对14个小麦属植物叶绿体基因组的倒位和基因重排进行分析(图2)。结果发现,相对于烟草,所有小麦属植物叶绿体基因组发生了两个明显的倒位事件,在LSC区段的psbD-trnfM-CAU间发生了一个大的倒位,而在这个大的倒位中psbD-trnT-GGU又发生了小的倒位。这两个倒位事件在所有植物叶绿体中均存在[13],说明这两个倒位事件在叶绿体进化过程中具有重要作用。在14个小麦属物种间,基因组的结构及基因排列顺序基本一致,没有明显的基因重排现象发生。
2.4 系统发育分析
以二穗短柄草为外源,对14个小麦属植物的叶绿体基因组进行全局比对,构建系统发育进化树(图3)。从图中可以看出,具有AAGG基因型的提莫菲维小麦单独聚为一支,其他13个小麦属植物具有AABB基因型的小麦属物种聚为另外一大支,反应了其系统进化关系。在这大分支中,圆锥小麦和斯卑尔脱小麦聚为1个小分支,其他11个小麦属植物聚为另一小分支,其中马卡小麦的3个种(T.macha.megrelicum、T.mach.palaeoimereticum和T.macha.Colchicum)聚在一起,但是其支持率只有54.757%,表明它们间存在较明显序列分化。圆锥小麦与斯卑尔脱小麦两个物种聚为一支,支持率为96.477%,明显高于其他物种,说明两者亲缘关系很近。

以烟草叶绿体基因组为参考。
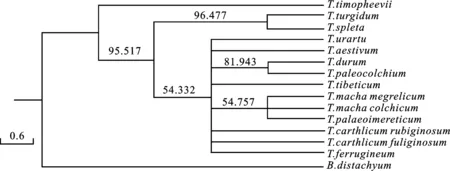
图3 基于叶绿体全基因组对14个小麦属植物进行的系统发育进化分析
2.5 RNA编辑分析
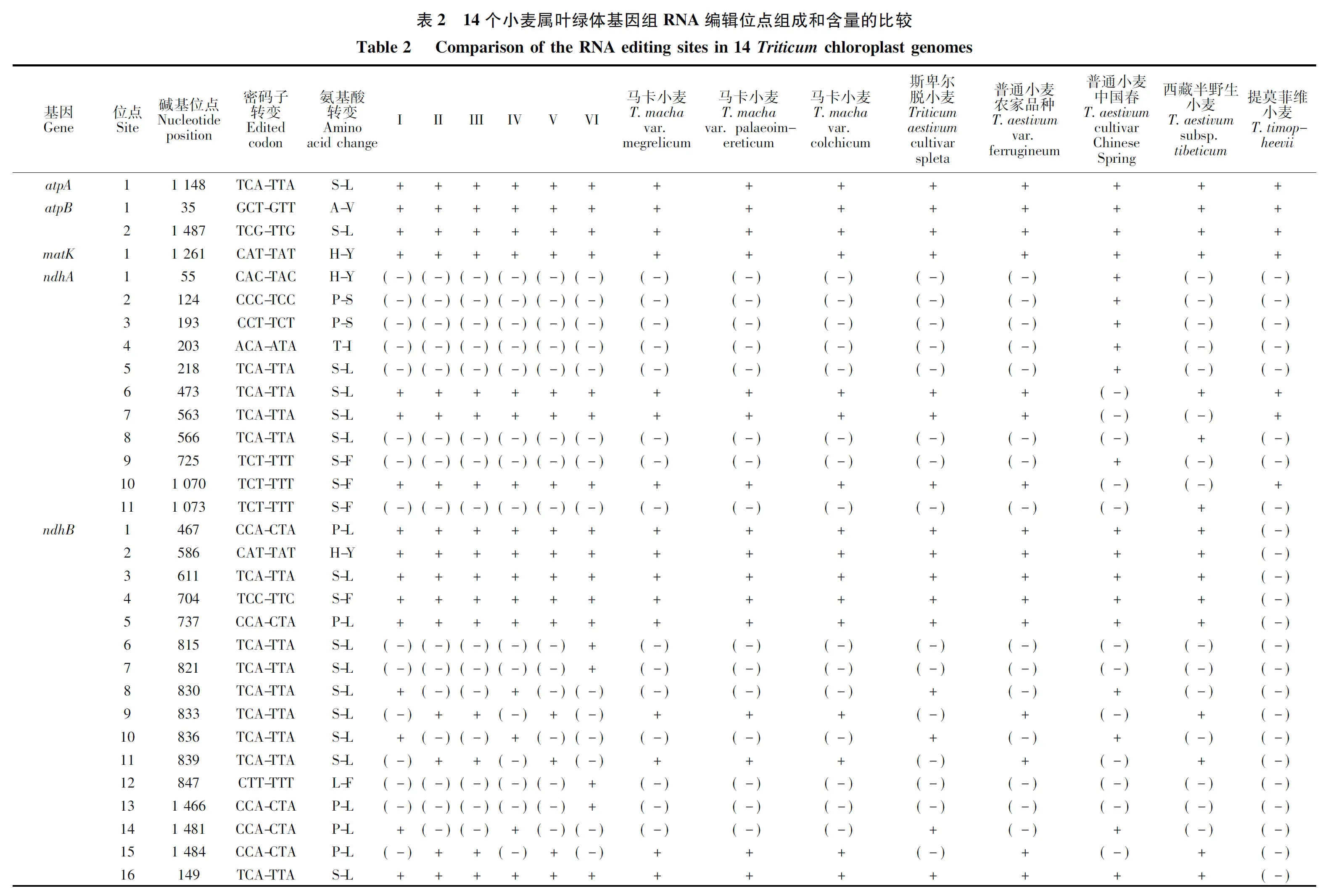
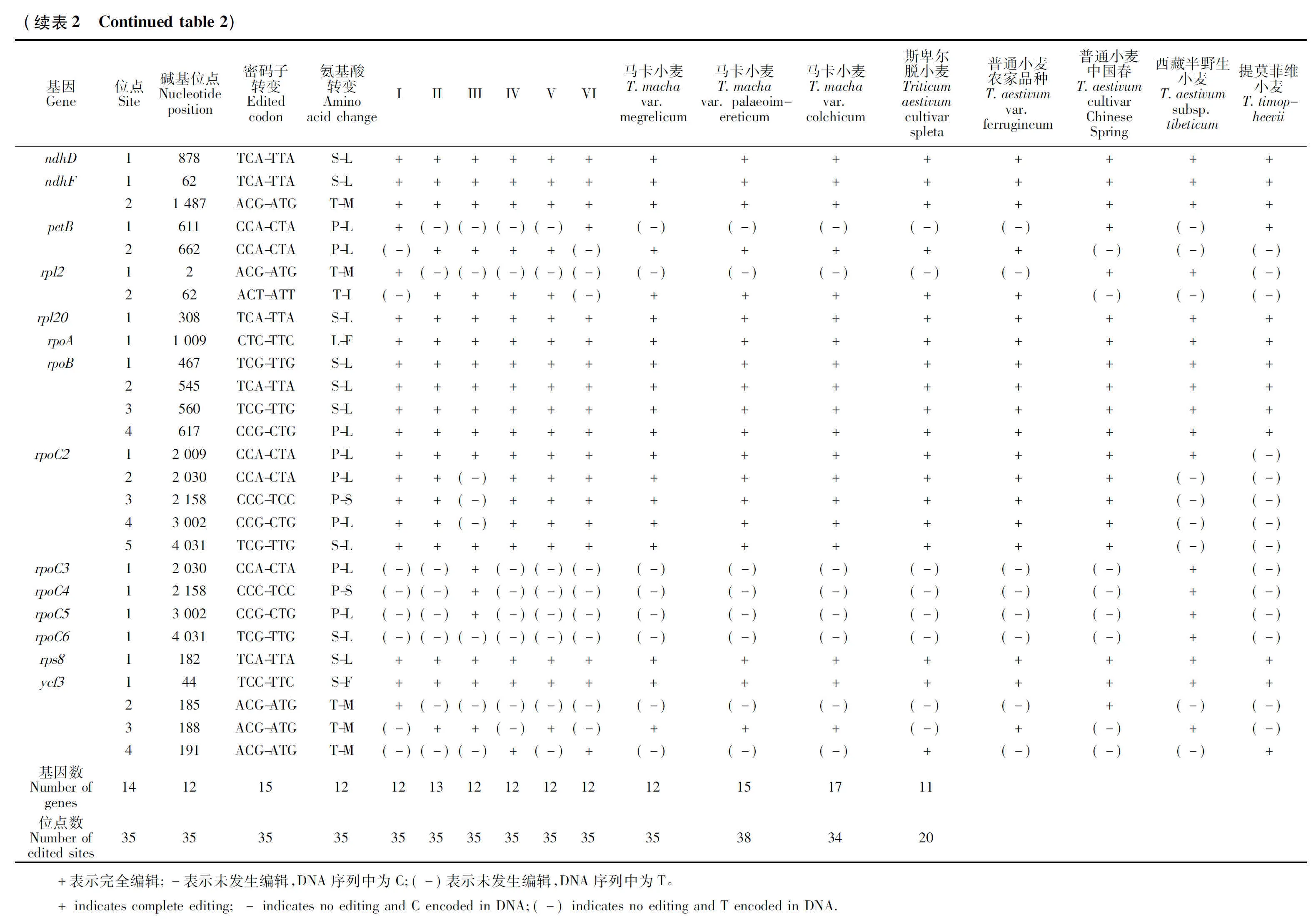
通过对14个小麦属植物叶绿体基因组的RNA编辑位点进行预测,共发现分布于19个基因上的58个RNA编辑位点(表2)。所有预测的编辑位点均为胞嘧啶(C)到胸腺嘧啶(T)的转换,且所有的编辑均引起了氨基酸的变化。其中基因ndhB的编辑位点数量最多,达16个;其次是ndhA(11个)、rpoC(5个)、rpoB(4个)、ycf3(4个)、atpB(2个)、ndhF(2个)、petB(2个)、rpl2(2个),其余基因均只有1个编辑位点。进一步分析发现,发生编辑的位点中,有9个位点位于密码子的第一位,剩余的都发生在密码子的第二位,没有发现密码子第三位发生编辑的位点。比较分析发现,小麦属植物叶绿体编辑位点具有较强的保守性,其中10个基因中的15个位点,包括atpA、atpB、matK、ndhD、ndhF、rpl20、rpoA、rpoB、rps8、ycf3,在所有小麦属物种中均发生了编辑。但提莫菲维小麦RNA编辑情况与其他物种差异较大,只在11个基因中预测到了20个编辑位点,尤其在ndhB和rpoC2处,RNA编辑位点明显较其他物种少。本研究也预测到了部分物种特异的RNA编辑位点,这为从叶绿体RNA编辑角度研究小麦属物种的起源进化提供了有益信息。
3 讨 论
研究发现,14个小麦属植物叶绿体的基因组大小在135 897~136 157 bp,各物种间差异不到1 000 bp,IR区长度在21 541~21 566 bp,与被子植物叶绿体基因组特征相吻合。进一步分析发现,小麦属植物叶绿体的LSC区长度的变异大于SSC和IR区,基因组长度的变异主要是LSC区长度的差异引起的。大多数小麦属植物叶绿体基因组的总基因数为105个,rRNA编码基因数物种间不存在差异,蛋白质编码基因在不同物种中基本一致,总基因数的差异主要是由tRNA编码基因数不同引起的。 虽然IR区段长度变异不大,但IR段基因数目存在较明显的变异,尤其是二粒系小麦的3个物种硬粒小麦、科尔希小麦和提莫菲维小麦IR区基因数目明显高于其他物种,说明二粒系小麦IR区较其他物种的遗传差异大,变异明显,这可能与二粒系小麦的遗传多样性相关。二粒系小麦中的提莫菲维小麦属于AAGG基因型,与AABB基因型的系统进化关系较远,IR区的变异程度也反映了其系统进化关系。叶绿体基因组上存在较高的基因转换能力,确保了2个IR序列的一致与稳定[14],选取的14个物种IR区长度基本一致,并未发生像天竺葵、豌豆、蚕豆等植物中IR过长、过短甚至完全丢失的情况[15]。
高等植物叶绿体基因组的4个边界发生扩张与收缩,是一个普遍的进化现象[16]。小麦属植物叶绿体基因组在LSC-IRb、SSC-IRa、IRa-LSC边界几乎不存在变异,最多有几个碱基的差异,只有硬粒小麦和乌拉尔图小麦在IRB-SSC边界基因与其他小麦属植物存在明显差异。MAUVE分析基因组结构重排和倒位也发现小麦属叶绿体基因组都呈线性化排列,基因组中的同源基因集簇重合度高,较为保守,各物种间基因的排列顺序基本一致且没有大的基因重排现象。基于全基因组序列比对分析,发现不同小麦属物种的基因间隔区序列差异大于基因编码区,其中psbI-psbD、rbcL-psaI、rpl22-rps19表现出较大的序列变异,可作为新的潜在序列资源用于小麦属物种的分子鉴定与系统进化研究。基于全叶绿体基因组的系统进化分析表明小麦属物种是单一起源的,并且AAGG基因型的提莫菲维小麦与其他AABB基因型的物种完全分开,基本反映了其系统进化与亲缘关系,表明叶绿体全基因组序列是研究小麦属物种的亲缘关系和进化提供重要的工具。
RNA编辑作为高等植物细胞器基因组转录后水平基因表达调控的一种重要方式,在植物生长发育过程中发挥着重要的作用[17]。目前已知除地钱外,所有陆生植物都存在RNA编辑现象[18]。RNA编辑具有双重功能,一是丰富蛋白质的多样性,二是维持种族发育的稳定性与保守性。由于RNA编辑位点突变比普通位点要快,mRNA 编辑可增加蛋白质的多样性,同时通过遗传变异又能维持一定的发育保守性,修复有害的基因组突变[19]。因此,RNA编辑可为揭示小麦属作物的起源与进化提供重要参考依据。通过对14个小麦属植物叶绿体基因RNA编辑位点的预测,发现有19个叶绿体基因发生了RNA编辑现象,共预测到58个RNA编辑位点。所有编辑位点均为胞嘧啶(C)到胸腺嘧啶(T)的转换,且所有的编辑均引起了氨基酸的变化,且编辑位点以发生在第二位密码子为主,少部分发生在密码子第一位,密码子第三位没有发现发生编辑的位点,这与前人发现的禾本科叶绿体基因编辑的特征一致[20]。ndhB基因上编辑位点最多,所有编辑位点碱基都是C→U的转变,编辑产生S→L的氨基酸转变形式最多,这与前人在禾本科的大麦[21]、玉米[22]上的研究结果一致,说明ndhB基因对植物生理生化过程具有重要调控作用。提莫菲维小麦叶绿体RNA编辑情况比其他小麦属物种存在更大变异,特别是在ndhB和rpoC2基因上没有发生编辑现象,这可能是该物种在长期进化过程中对环境适应性变化或者物种特异性造成的,对其特异编辑位点生物学功能的研究将为揭示小麦属物种的起源、进化及相关分子机制提供重要信息。
猜你喜欢
军事文摘(2022年16期)2022-08-24
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
支部建设(2020年15期)2020-07-08
课程教育研究·学法教法研究(2019年18期)2019-10-08
少儿科技(2019年9期)2019-09-10
江苏农业科学(2016年2期)2016-04-11
百科知识(2015年18期)2015-09-10
小星星·阅读100分(高年级)(2015年4期)2015-05-26
