
专一性酶解-高效液相色谱-荧光检测法测定3种食用油中4种多环芳烃
2020-07-06 06:01:48章再婷王永健王春芳陈树兵曹国洲
理化检验-化学分册 2020年6期
章再婷,杨 燕,王永健,王春芳∗,李 双,陈树兵,曹国洲
(1.宁波中盛产品检测有限公司,宁波 315012;2.宁波海关技术中心,宁波 315012)
多环芳烃(PAHs)作为一类强致癌性化合物,主要来源于反复使用或高温油炸的非正常食用油中[1-2]。苯并[a]蒽(Ba A)、苯并[b]荧蒽(BbF)、苯并[k]荧蒽(Bk F)和苯并[a]芘(BaP)是毒性较强的4种PAHs,其中BaP被国际癌症研究机构归为1类致癌物,其余3种被归为2B 类致癌物[3-5]。受暴利的驱使,非正常食用油越来越频繁的出现在人们的生活中,对人们的身体健康造成巨大的威胁,是国家严厉打击的食品违法行为的对象。因此,建立能够快速同时测定食用油中这4 种PAHs含量的方法尤为重要。
目前建立的食用油中单一或多个PAHs的快速筛查方法主要涉及两方面内容:前处理方法和仪器分析方法。仪器分析方法包括液相色谱-荧光检测法(LC-FLD)、气相色谱-氢火焰离子化检测法(GC-FID)、气相色谱-质谱法(GC-MS)和液相色谱-质谱法(LC-MS)等[6-13]。其中,GC-MS 和LC-MS对前处理净化条件要求比较严格,油脂的离子化抑制效应明显,其回收率及稳定性均劣于LC-FLD 和GC-FID 的。由于4 种上述PAHs均具有多环结构,LC-FLD 的专一性更强,其对加工处理的或按照比例渗入的食用油的检出限均能达到ng·kg-1级,更适用于食用油中微量化合物的鉴别分析。前处理方法主要包括基质分散萃取法(Qu ECh ERS等),固相萃取法(HLB小柱、中性氧化铝柱等)和凝胶渗透色谱法(GPC)等[6-13]。其中固相萃取法中是通过氢键作用将待测物保留在吸附剂上,其虽能有效除去油脂基质,但操作过程繁琐耗时、重现性差。文献[8]建立了地沟油中BaP含量检测的Oasis HLB小柱净化方法,但该方法对于含有多个环状结构的亲脂性待测物的回收率不高;GPC虽具备自动化优势,但单样净化耗时较长(3 h),且消耗有机溶剂较多,试验成本高。油脂中亲脂性待测物的快速、高效提取,是食用油中PAHs含量检测面临的一个艰巨挑战。
目前,脂肪酶酶解技术主要应用于营养成分分析,如脂肪酸及各种维生素的检测,在食品添加剂及有毒有害物质检测方面应用甚少。本工作根据这4种PAHs的多环结构和食用油基质中高脂肪含量的特点,前处理采用专一性酶解法处理样品,采用高效液相色谱-荧光检测法(HPLC-FLD)对样品提取液中Ba A、Bb F、Bk F 和BaF 等4种PAHs的含量进行了测定,以期为提高政府监管部门的监管工作效率,保证食品安全提供了基础。
1 试验部分
1.1 仪器与试剂
LC-20AT 型高效液相色谱仪,配荧光检测器;Milli-Q 型高纯水发生器;SF-40R 型冷冻离心机;TKA 型漩涡振动器;Hei-VAP Expert型旋转蒸发仪;DK-S28型电热恒温水浴锅;0.22μm 尼龙滤膜。
单标准储备溶液:取0.01 g单标准品,用二氯甲烷溶解,转移至10 mL 的容量瓶中,用乙腈稀释至刻度,得到1.0 g·L-1的单标准储备溶液,于-20 ℃下保存。
磷酸盐缓冲溶液:0.108 g·mL-1,称取54 g的磷酸二氢钾,用500 mL 水溶解,氢氧化钾调至p H为8.0。
混合标准溶液:称取4种PAHs的单标准储备溶液各10μL,用乙腈定容至10.0 mL,配制成1.0 mg·L-1标准溶液,于-20 ℃避光保存。
混合标准溶液系列:称取适量混合标准溶液,用乙腈稀释至刻度,配制0,0.1,0.5,1.0,2.0,5.0μg·L-1的混合标准溶液系列。
Ba A、Bb F、Bk F 和BaF 的纯度不小于95%;脂肪酶的酶活性大于700 U·mg-1。
正己烷、乙腈、二氯甲烷、乙醇均为色谱纯;其他试剂为分析纯;试验用水为超纯水(电阻率18.2ΩM·cm)。
动物油、植物油和煎炸油等3种常见食用油来自国家残留监控抽样和进出口送检企业。
1.2 仪器工作条件
Dikma Plus C18色谱柱(250 mm×4.6 mm,5.0μm),柱温40 ℃;流动相:A 为水,B 为乙腈;流量1.0 mL·min-1;进样量20μL;梯度洗脱程序见表1。0~10 min时,Ba A 的荧光激发波长(λex)为270 nm,发射波长(λem)为380 nm;10~20 min时,Bb F、Bk F和BaP的λex为294 nm,λem为406 nm。
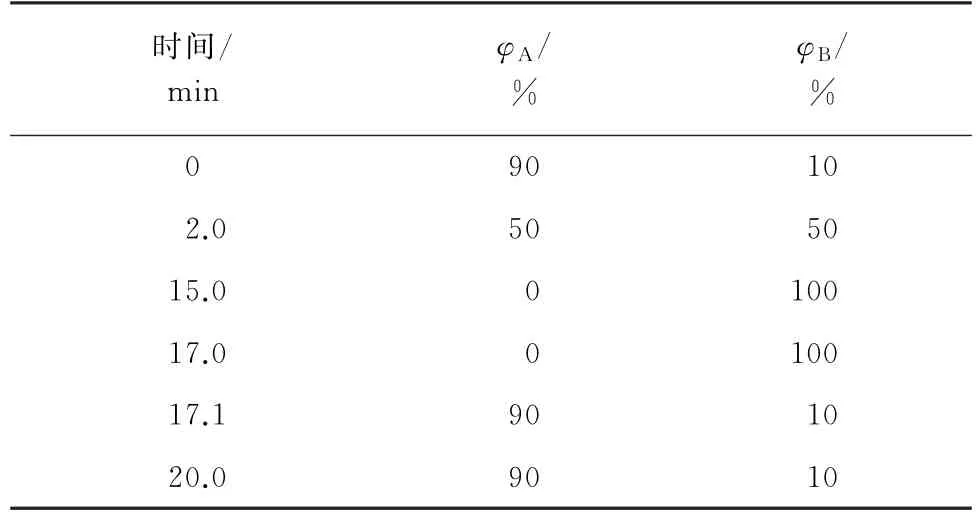
表1 梯度洗脱程序Tab.1 Program of gradient elution
1.3 试验方法
称取2.0 g样品,依次加入5 mL磷酸盐缓冲溶液(pH 8.0)和0.2 g脂肪酶,恒温(37±2)℃振荡酶解2 h。加入1.0 g碳酸钾,5 mL 乙醇,涡旋混匀。在提取溶液中加入15 mL 正己烷,5 mL 水,振荡5 min,以4 500 r·min-1转速离心5 min,取下层水相按上述步骤重复萃取一次。合并萃取液,于40 ℃旋蒸至干,用1.0 mL乙腈复溶,过0.22μm 滤膜,滤液按照仪器工作条件进行测定。
2 结果与讨论
2.1 色谱行为
按照仪器工作条件,对5.0μg·L-1混合标准溶液进行测定,色谱图见图1。Ba A、BbF、Bk F和BaP的保留时间分别为9.3,12.0,12.6,13.9 min。

图1 4种PAHs混合标准溶液的色谱图Fig.1 Chromatogram of mixed standard solution of the 4 PAHs
2.2 脂肪酶、酶解时间及碳酸钾用量的选择
针对食用油特殊基质成分和4 种PAHs的弱极性性质,试验采用专一性脂肪酶处理样品,缓冲溶液选择磷酸二氢钾缓冲溶液(pH 8.0),整个酶解过程温度控制在(37±2)℃,此条件可以在保证酶活性的同时,又可以降低基质中脂肪的干扰。皂化反应经常用来去除油脂,其通常在浓碱环境下进行(氢氧化钾或氢氧化钠等强碱性化合物),但在此环境中,待测化合物的结构极易被破坏,且有机相中残留的强碱性化合物需多次水洗才可以消除,不可避免的造成待测化合物的损失、检测时间的加长和检测成本的增加。因此,本工作采用碳酸钾进行皂化反应,碳酸钾不仅可以提供弱碱性环境和终止酶解反应,还可以皂化油脂,确保最大的除油率,除油率(X)可通过测量氮吹充分后残余油脂的精确质量值计算:

式中:m X是指经脂肪酶水解和碳酸钾皂化除油后的残余油脂质量,g;m0是指未经任何处理的残余油脂质量,g;X,%。
脂肪酶用量以及酶解时间都与除油率相关联。试验考察了脂肪酶用量分别为0.05,0.1,0.2,0.5,1.0 g,酶解时间分别为1.0,2.0,3.0,5.0 h时对动物油、植物油和经用于煎炸的食用油(煎炸油)等3种常见食用油除油率的影响。
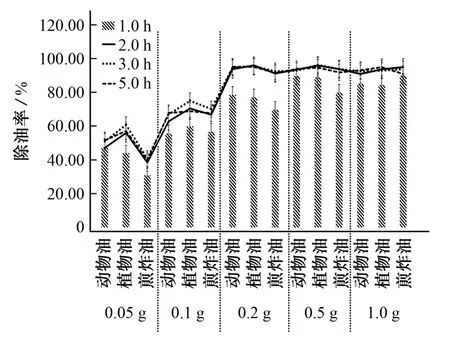
图2 脂肪酶用量、酶解时间对3种食用油除油率的影响Fig.2 Effect of the amount of lipase and time of enzymolysis on the rate of oil-removal of the 3 edible oils
由图2可知:当脂肪酶用量为0.05 g和0.1 g时,除油率受酶解时间影响较大,此时,酶解时间大于等于2.0 h的除油率较1.0 h的明显增加,但均低于80%;当脂肪酶用量大于0.1 g 时,酶解时间2.0 h的除油率明显高于1.0 h的,但2.0,3.0,5.0 h的除油率差别很小。从节约时间和成本的角度出发,试验选择脂肪酶用量为0.2 g,酶解时间为2.0 h。
试验还考察了碳酸钾用量分别为0,0.5,1.0,2.0 g时对除油率的影响。
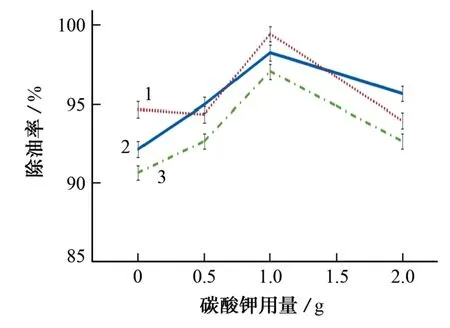
图3 碳酸钾用量对3种食用油除油率的影响Fig.3 Effect of the amount of K2 CO3 added on the rate of oil-removal of the 3 edible oil samples
由图3可知:当碳酸钾的用量不大于1.0 g时,除油率随着碳酸钾用量的增加而增加;当碳酸钾用量为1.0 g 时,除油率达到最大区间(98.1%~99.8%);碳酸钾用量为2.0 g时,除油率反而降低,推测可能是由于过量的碳酸钾造成了过碱环境,破坏了酶的活性进而降低了除油效率。因此,试验选择碳酸钾的用量为1.0 g。
2.3 乙醇和正己烷用量的选择
萃取前,在待皂化的样品溶液中加入适量乙醇可以有效防止乳化现象的发生,此外,乙醇还能起到预萃取的作用。同时,4种PAHs属于弱极性化合物,萃取剂正己烷的用量对其回收率的影响较大。因此,试验考察了乙醇用量分别为2.5,5,10,15 mL,正己烷用量分别为15,30,45 mL时对混合标准溶液的添加量为0.1μg·kg-1的动物油、植物油和煎炸油等3类常见食用油中4种PAHs回收率的影响,试验发现乙醇用量为2.5 mL时,体系乳化现象仍严重,无法计算回收率,其余结果见表2~表5。
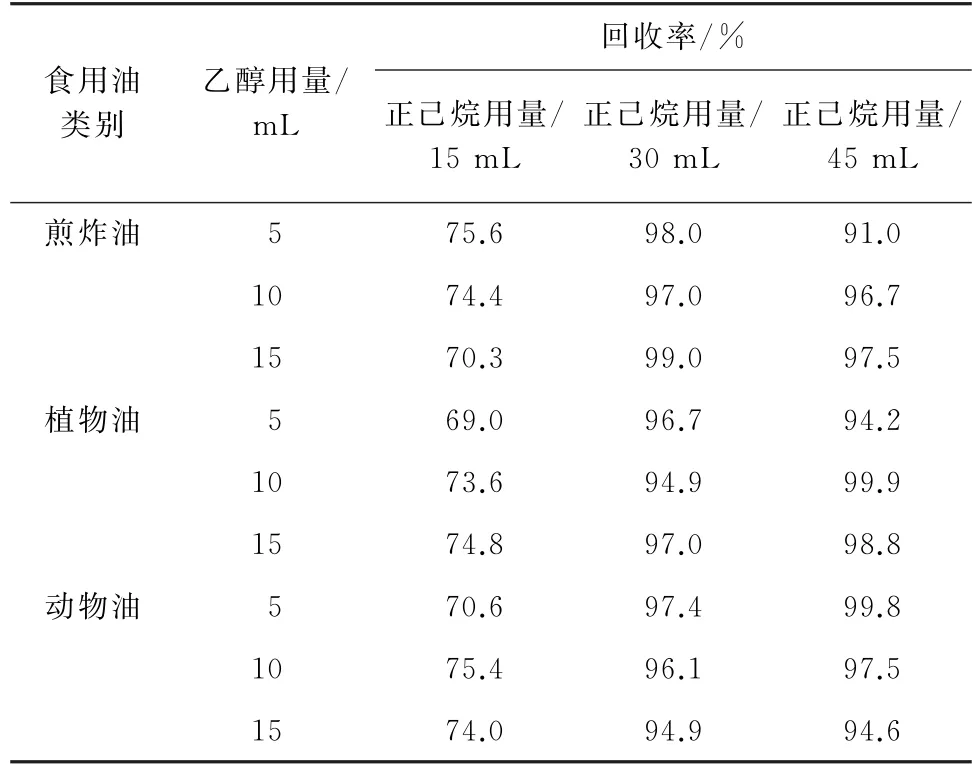
表2 不同乙醇和正己烷用量下3种常见食用油中BaA回收试验结果Tab.2 Results of test for recovery of BaA in the 3 edible oils with different amounts of ethanol and n-hexane
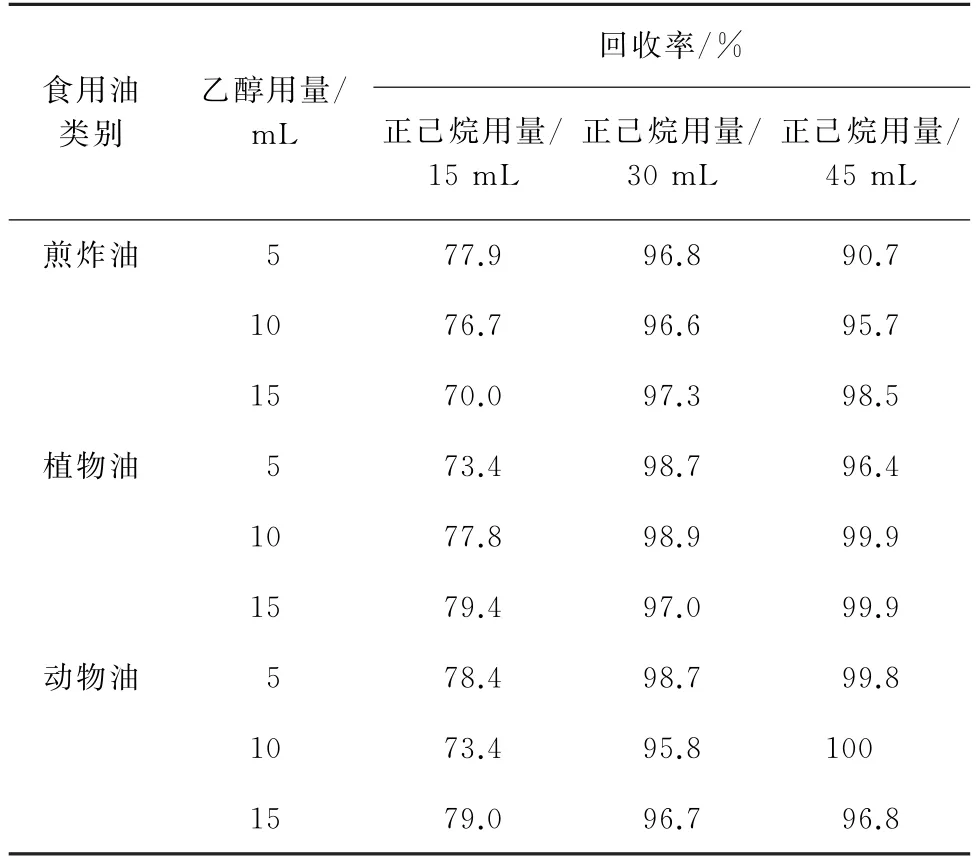
表3 不同乙醇和正己烷用量下3种常见食用油中BbF回收试验结果Tab.3 Results of test for recovery of BbF in the 3 edible oils with different amounts of ethanol and n-hexane
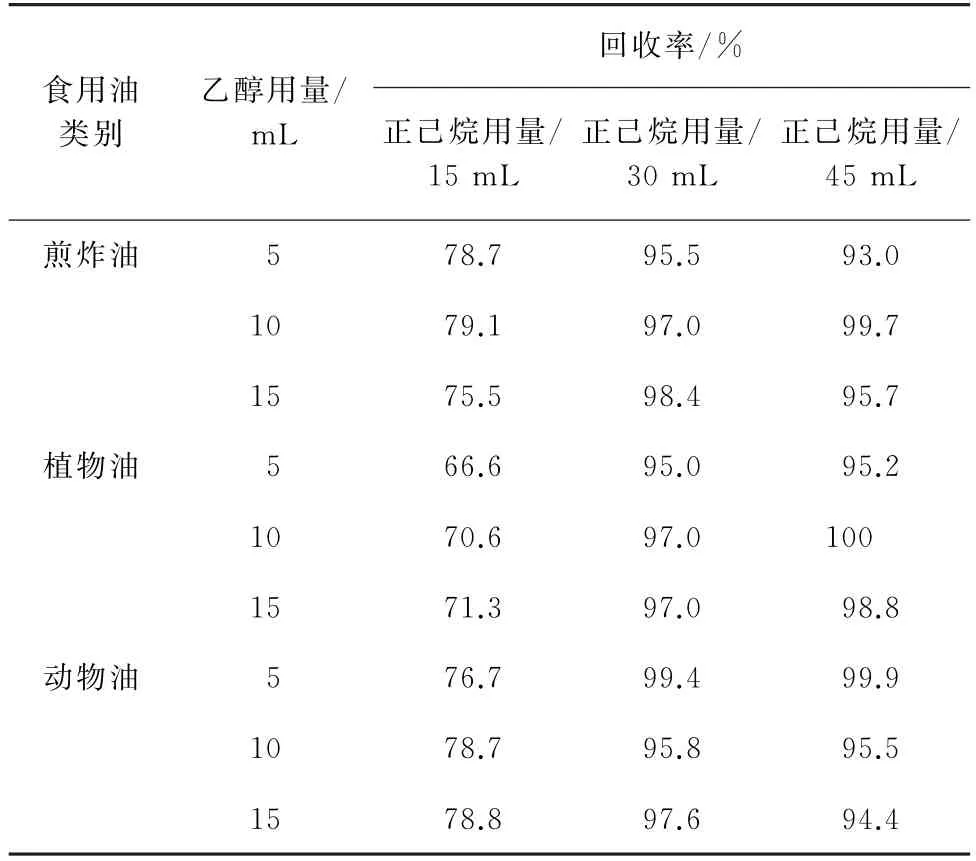
表4 不同乙醇和正己烷用量下3种常见食用油中BkF回收试验结果Tab.4 Results of test for recovery of Bk F in the 3 edible oils with different amounts of ethanol and n-hexane
由表2~表5结果可知:正己烷用量为15 mL时,4种PAHs的回收率较低,均低于77%;当正己烷用量为30 mL和45 mL 时,回收率趋于稳定,均能达到90%以上。当乙醇用量不小于5 mL 时,4种PAHs回收率相差不大,而乙醇用量为5 mL时即可保持液-液界面清晰消除乳化现象。从节约浓缩时间和成本的角度出发,试验选择的乙醇用量为5 mL,正己烷用量为30 mL。

表5 不同乙醇和正己烷用量下3种常见食用油中BaP回收试验结果Tab.5 Results of test for recovery of BaP in the 3 edible oils with different amounts of ethanol and n-hexane
2.4 流动相及色谱柱的选择
采用液相色谱检测目标化合物时,经常在流动相中加入缓冲盐以增加仪器的响应灵敏度,但缓冲盐的大量长期使用会对仪器造成损害。试验考察了分别以甲醇-水和乙腈-水体系作流动相时对4 种PAHs的色谱行为的影响。结果表明:乙腈-水体系柱压更低、安全性好,所以,试验选择乙腈-水体系为流动相,通过对其梯度洗脱条件的优化,得到了合适的保留时间、峰形及分离效果。另外,试验还对Dikma Plus C18色谱柱和Hypersile Gold C18色谱柱的分离效果进行了考察,结果显示,2种色谱柱仅表现出组分保留时间的差异,分离度均可满足检测需要,考虑到经济成本,试验选择了Dikma Plus C18色谱柱。
2.5 标准曲线、检出限和测定下限
按照试验方法对混合标准溶液系列进行测定,以保留时间定性、外标法定量。以4种PAHs的质量浓度为横坐标,其对应的仪器响应值(响应值=标准物质的峰面积/标准物质的质量浓度为纵坐标绘制工作曲线。4 种PAHs标准曲线的线性范围为0.1~5.0μg·L-1,线性回归方程、相关系数见表6。
以3倍信噪比和10倍信噪比计算检出限(3S/N)和测定下限(10S/N),结果见表6。
2.6 精密度和回收试验
在3种常见食用油中进行3个浓度水平的加标回收试验,计算回收率和测定值的相对标准偏差(RSD),结果见表7。
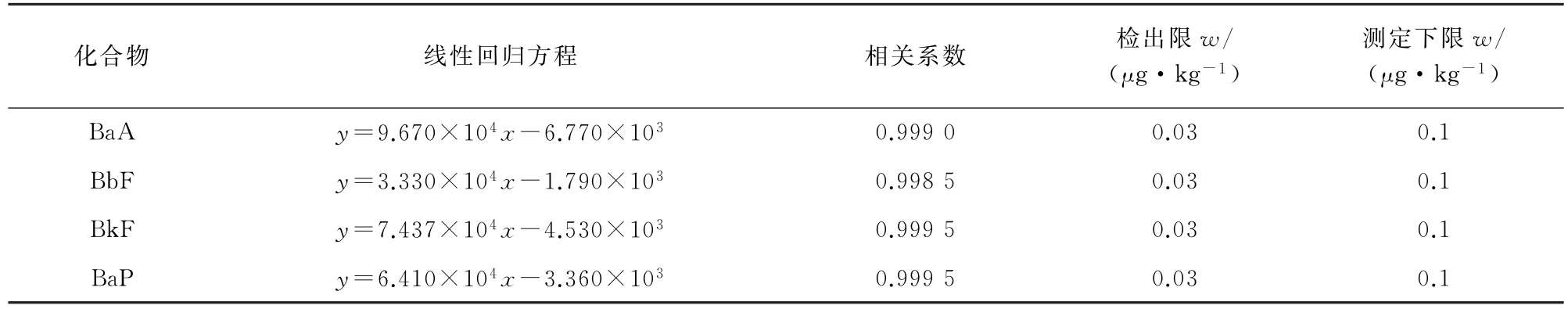
表6 线性参数、检出限和测定下限Tab.6 Linearity parameters,detection limits and lower limits of determination
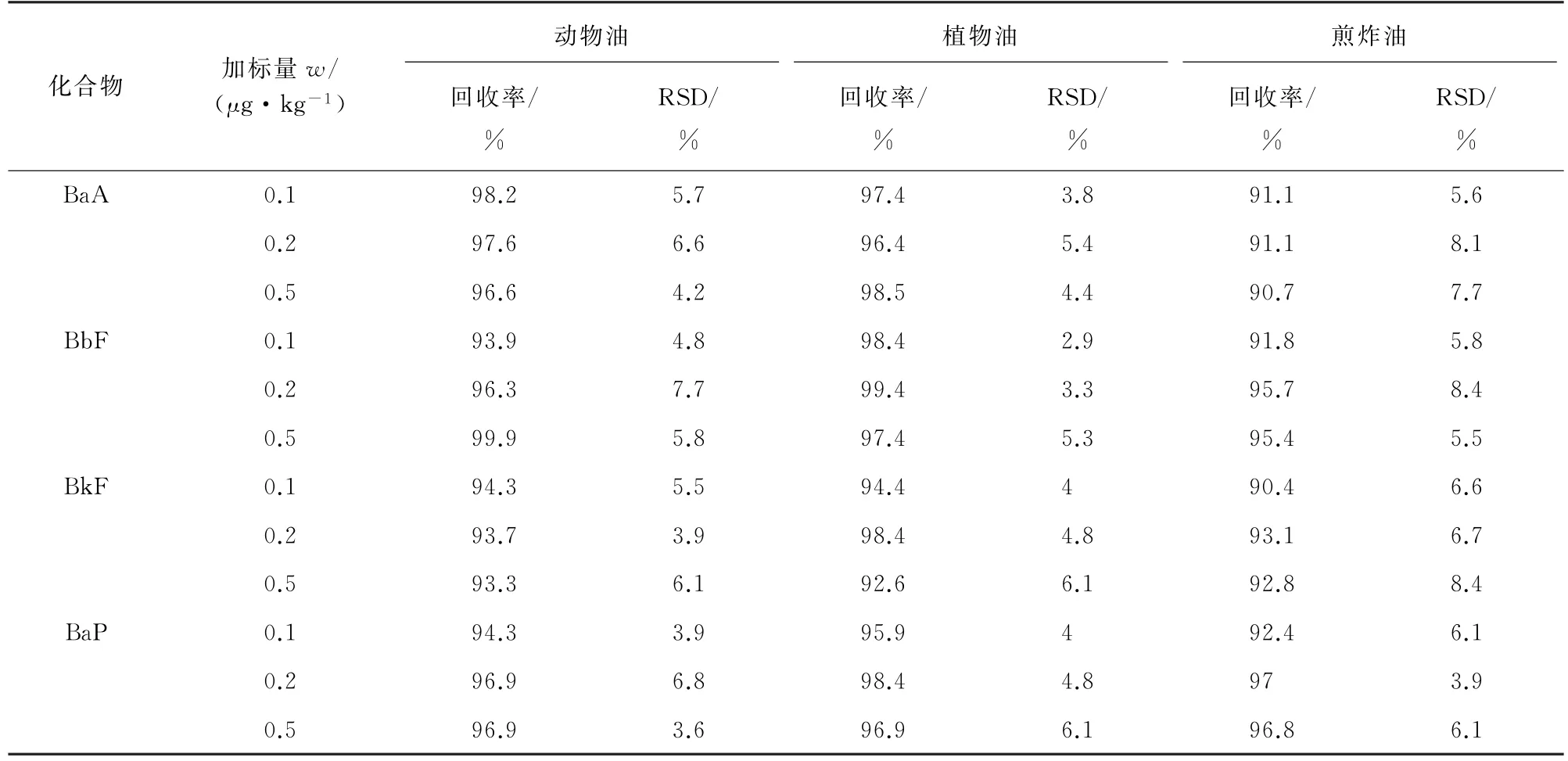
表7 精密度和回收试验结果(n=5)Tab.7 Results of tests for precision and recovery(n=5)
由表7可知:4种PAHs的回收率为90.4%~99.9%,RSD 均在10%以内,说明本方法对这3种食用油均具有较强的适用性,可实现油脂中4 种PAHs的快速提取、净化的目的。
本工作根据4种PAHs的多环结构以及食用油基质中高脂肪含量的特点,建立了专一性酶解-高效液相色谱-荧光检测法测定3 种食用油中4 种PAHs含量的方法,该方法大大降低了前处理时间和油脂的基质抑制效应,检出限远低于现有方法[6-11],为各类食用油中这4种PAHs的检测开辟了新的途径,可作为油脂中亲脂性化合物的快速检测技术广泛推广。
猜你喜欢
河南畜牧兽医(2021年9期)2021-12-10 10:43:50
应用化工(2021年2期)2021-03-12 10:10:42
石油沥青(2020年1期)2020-05-25 06:54:04
杭州化工(2020年1期)2020-05-09 14:00:54
山东冶金(2019年6期)2020-01-06 07:46:02
伴侣(2019年10期)2019-10-16 02:23:34
中国油脂(2019年8期)2019-08-22 10:25:50
今日农业(2019年12期)2019-08-15 00:56:32
中国油脂(2019年3期)2019-04-29 01:28:34
今日农业(2019年14期)2019-01-04 08:57:40
