
基于β-二酮配体的三价铕配合物合成与表征
2020-07-01 09:53郑昕怡杨欣瑜王依婷吴姣姣
湖州师范学院学报 2020年4期
郑昕怡, 杨欣瑜, 王依婷, 吴姣姣, 陈 超
(湖州师范学院 生命科学学院, 浙江 湖州 313000)
在有机发光材料中,红色是最薄弱的一环,这是因为红色发光的电子跃迁能隙较小,很难与载流子传输层能量匹配,不能使电子与空穴有效地在发光区域结合.1942年Weissman 发现某些β-二酮类的三价铕配合物可以用近紫外光激发,发出明亮的红光[1-2].至此,对这类配合物的合成和发光性质的研究引起了众多科研者的兴趣[3-7].稀土Eu(Ⅲ)离子独特的电子结构决定了它特殊的光学性能,而通过配体和稀土离子的相互作用及影响,又可以极大地改变、修饰和增强这些特性[8-9].如果进行特殊的分子设计还可以有效地改善稀土铕配合物的发光性能,这对研究红色发光材料具有重要意义[10-12].本文以β-二酮2-噻吩甲酰三氟丙酮(HTTA)和4,4,4-三氟-1-苯基-1,3-丁二酮(HBTA)为第一配体,三苯基氧膦(TPPO)为第二配体,分别与六水合三氯化铕(EuCl3·6H2O)反应,合成[Eu(TTA)3(TPPO)2](简称铕配合物1)和[Eu(BTA)3(TPPO)2](简称铕配合物2)两个三价铕配合物,通过氢核磁共振、红外光谱、紫外光谱和X-射线单晶衍射对其结构进行表征,并研究它们的荧光性质.
1 实验材料与方法
1.1 实验试剂
2-噻吩甲酰三氟丙酮(HTTA)、4,4,4-三氟-1-苯基-1,3-丁二酮(HBTA)、三苯基氧膦(TPPO)、六水合三氯化铕(EuCl3·6H2O)(上海阿拉丁生化科技股份有限公司,分析纯)、无水乙醇、氢氧化钠(国药集团化学试剂有限公司,分析纯).
1.2 铕配合物1和2的合成
铕配合物1和2的合成方法如图1所示[13].将第一配体HTTA或HBTA(3 mmol)和氢氧化钠(120 mg,3 mmol)加入到100 mL圆底烧瓶中,再加入15 mL乙醇和1.5 mL去离子水,室温下搅拌15 min使其溶解.然后将六水合三氯化铕(366 mg,1 mmol)溶于1 mL去离子水,缓慢滴加到反应瓶中,滴加完毕后加热至80 ℃继续搅拌30 min.再将第二配体TPPO(556 mg,2 mmol)溶于20 mL乙醇中,缓慢滴加到反应瓶中,反应过程中析出大量的不溶物质.反应8 h后趁热抽滤,得到的固体用10 mL无水乙醇洗涤3次,烘干,得到铕配合物1(白色粉末926 mg,产率为84%)和铕配合物2(白色粉末643 mg,产率为81%).

2 结果与讨论
2.1 铕配合物1和2氢核磁共振(1H NMR)表征
反应分离得到的铕配合物1经1H NMR表征如图2(a)所示,第一配体2-噻吩甲酰三氟丙酮分子(HTTA)在碱的作用下脱去一个质子,从二酮结构变为烯醇负离子结构(TTA),其中2-位CH信号以单峰形式出现在3.74 ppm处,噻吩基团的3个特征峰(CH)分别以单峰的形式出现在7.46、6.51和6.33 ppm处,4个信号峰的积分比为1∶1∶1∶1;第二配体三苯基氧膦(TPPO)芳环上的氢核磁信号出现在7.57~7.69 ppm处,符合苯环上氢核磁信号峰的特征.将核磁信号峰积分后,氢总数为42,符合铕配合物1的分子式C44H42P2O8S3F9中氢的个数,第一配体(TTA)和第二配体(TPPO)比值为3∶2.氢谱数据1H NMR(400,MHz,DMSO-d6)为:δ7.57~7.69(m,30H),7.46(s,3H),6.51(s,3H),6.33(s,3H),3.74(s,3H).
铕配合物2的氢核磁共振数据(图2(b))所示,第一配体4,4,4-三氟-1-苯基-1,3-丁二酮(HBTA)同样在碱的作用下脱去一个质子,从二酮结构变为烯醇负离子结构(BTA),其中2-位CH信号以单峰形式出现在4.19 ppm处,第一配体BTA芳环上的氢核磁信号峰和第二配体TPPO芳环上的氢核磁信号峰出现在7.19~7.68 ppm处,符合苯环上氢核磁信号峰的特征.将核磁信号峰积分后,氢总数为48,符合铕配合物2的分子式C66H48P2O8F9中氢的个数,第一配体(BTA)和第二配体(TPPO)的比值为3∶2.氢谱数据1H NMR(400 MHz,DMSO-d6)为:δ7.63~7.68(m,17H),7.56~7.59(m,12H),7.19~7.25(d,16H),4.19(s,3H).

2.2 铕配合物1和2的红外光谱(IR)表征
铕配合物1和2的红外光谱图如图3的(a)、(b)所示,在1 610 cm-1和1 603 cm-1处出现的信号峰分别归属于铕配合物1和2中β-二酮配体C=O羰基键的伸缩振动峰,而在1 535 cm-1、1 549 cm-1左右处出现很强的C=C双键的伸缩振动峰,表示β-二酮配体以烯醇式负离子结构发生配位时的特征光谱峰,这也证实在合成的配合物中铕(Ⅲ)离子与配体的氧原子发生了配位[7-9].位于1 173 cm-1、1 187 cm-1处的信号峰分别归属于铕配合物1和2中第二配体TPPO的P=O双键的伸缩振动峰;1 455 cm-1、1 413 cm-1处分别为铕配合物1和2中C-P键的伸缩振动峰;3 050 cm-1、3 120 cm-1处分别出现了铕配合物1和2中配体TPPO的C-H的伸缩振动峰,说明第二配体TPPO也参与了配位;位于3 447 cm-1、3 449 cm-1处的强吸收峰则是这两个铕配合物所含水形成的氢键特征峰.

2.3 铕配合物1和2的紫外光谱(UV)表征
铕配合物1及其配体HTTA、TPPO的紫外光谱如图4(a)所示.由图4(a)可以看出,铕配合物1及其配体在紫外区均有较强的吸收,在350 nm处有一个强吸收峰.其配体HTTA在262 nm处有一个强吸收峰,与铕离子形成配合物后,其最大吸收峰蓝移了42 nm.配体TPPO在223 nm处有一个强吸收峰,但与铕离子形成配合物后,其最大吸收峰消失,这是由于配体与铕离子形成配合物后,产生更大的刚性共轭体系,改变了原有基团的吸电子及给电子能力,电子跃迁所需的能量减小,其223 nm处的最大吸收峰向可见光方向移动,故只显示一个吸收峰.此外,峰位及峰强度的改变也证明配体与稀土铕离子形成了键合作用.
铕配合物2及其配体HBTA、TPPO的紫外光谱如图4(b)所示.铕配合物2及其配体在紫外区也有较强的吸收,在324 nm处有强吸收峰.其配体HBTA在246 nm、326 nm处有强吸收峰,与铕离子形成配合物后,其吸收峰发生了小程度的蓝移.配体TPPO在223 nm处有一个最大的吸收峰,与铕离子形成配合物后,其吸收峰位置基本不变.比较铕配合物2及其配体HBTA、TPPO的紫外吸收峰后,发现配合物2的吸收基本是配体HBTA和TPPO的叠加,两种配体对能量吸收均有贡献.此外,峰位和峰强度的改变也证明配体与稀土铕离子形成了键合作用.

2.4 铕配合物1和2的晶体结构
金属配位化合物的晶体可通过多种方法获得[14].本文将铕配合物1用氯仿溶解和过滤后,将滤液(5 mL)转移到试管中,然后在氯仿上层小心地加上正己烷,静置培养晶体.通过正己烷的缓慢扩散,成功地得到了铕配合物1的晶体,经X-射线单晶衍射测试,其结构如图5(a)所示.在铕配合物1的结构中,中心Eu(Ⅲ)分别与三个2-噻吩甲酰三氟丙酮(TTA)配体的六个氧原子和两个三苯氧膦(TPPO)配体的两个氧原子配位,呈现八配位的多面棱镜形态.其中两个TTA配体的四个氧原子O(3)、O(4)、O(7)、O(8)和另外一个TTA配体的氧原子O(5)构成一个扭曲的配位平面,两个TPPO分别处于这个平面的两侧.Eu-O键的键长在2.358(3)~2.452(3) Å之间,处于正常的Eu-O键的键长范围内[4].TPPO配体与铕离子配位的Eu-O键的键长分别为2.358(3) Å和2.382(3) Å,两者相差不大.TTA配体与中心铕离子配位的Eu-O键的键长:靠近噻吩基团的Eu-O键的键长分别为2.452(3) Å、2.452(3) Å和2.450(3) Å;靠近三氟甲基的Eu-O键的键长分别为2.364(3) Å、2.359(3) Å和2.368(3) Å,后者的键长较前者短,说明靠近三氟甲基吸电子取代基的羰基在碱的作用下形成了烯醇负离子,并与铕离子配位,形成较强的离子型配位键.铕配合物1的部分键长(Å)和键角(deg)见表1.


表1 铕配合物1的部分键长(Å)和键角(deg)
铕配合物2晶体的培养方式与铕配合物1相同,晶体结构如图5(b)所示.在铕配合物2中也含有三个第一配体4,4,4-三氟-1-苯基-1,3-丁二酮(BTA)和两个第二配体三苯基氧膦(TPPO),呈现八配位的多面棱镜形态.其中两个BTA配体的四个氧原子O(3)、O(4)、O(5)、O(6)和另外一个BTA配体的氧原子O(2)构成一个扭曲的配位平面,两个三苯氧膦配体(TPPO)分别处于这个平面的两侧.与配合物1类似,在铕配合物2中,Eu-O键的键长在2.356(3)~2.463(2) Å之间,也处于正常的Eu-O键的键长范围内[4].TPPO配体与铕离子配位的Eu-O键的键长分别为2.385(3) Å和2.358(2) Å.BTA配体与中心铕离子配位的Eu-O键的键长:靠近苯基取代基的Eu-O键的键长分别为2.412(2) Å、2.435(2) Å和2.463(2) Å;靠近三氟甲基的Eu-O键的键长分别为2.376(3) Å、2.372(2) Å和2.356(3) Å,后者的键长较前者短,说明靠近吸电子取代基三氟甲基的羰基在碱的作用下形成了烯醇负离子,形成较强的离子型配位键.铕配合物2的部分键长(Å)和键角(deg)见表2.铕配合物1和2的晶体学数据见表3.
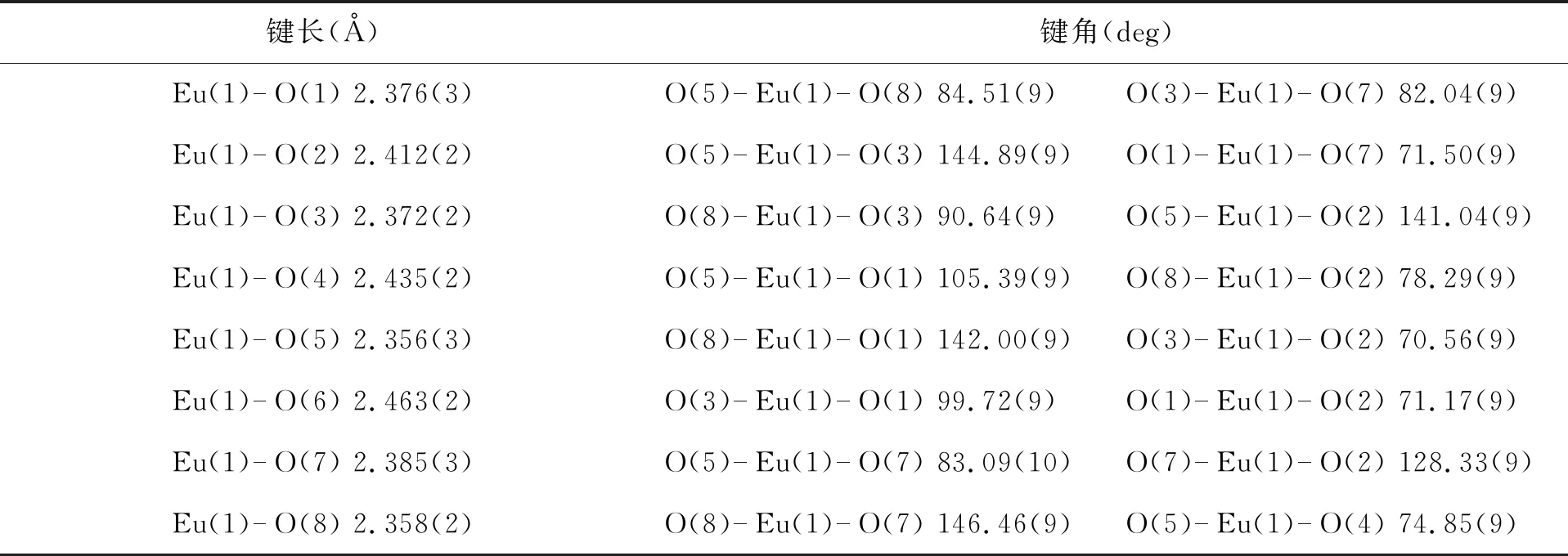
表2 铕配合物2的部分键长(Å)和键角(deg)Tab. 2 Selected bond lengths (Å) and bond angles (deg) of europium complex 2

表3 铕配合物1和2的晶体学数据Tab. 3 Crystallographic data of europium complexes 1 and 2
2.5 铕配合物1和2的荧光性质[15]
将铕配合物1和2分别溶于二甲亚砜,测试得到铕配合物1和2的激发光谱和发射光谱,见图6的(a)和(b).在激发光谱中,铕配合物1和2的最大激发波长分别为408 nm和398 nm,处于近紫外区域,属7F2→5D0能级跃迁;在465 nm处,铕配合物1和2均有一个较小的激发波,激发波长为7F1→5D0的能级跃迁;在536 nm处的激发波归属7F0→5D0的能级跃迁.在发射光谱中,铕配合物1和2的5D0→7F0、5D0→7F1和5D0→7F2的能级跃迁分别在537 nm、587 nm、615 nm、651 nm和702 nm处形成发射峰,这些发射峰为铕(Ⅲ)离子的特征荧光发射峰[16-17],其中以615 nm处的射峰为最强,说明铕配合物1和2以5D0→7F2能级跃迁为主.铕化合物不同发射峰的发射强度受其所处位置的对称性影响非常显著,当配合物中Eu(Ⅲ)离子周围环境不具有反演中心对称性时,以5D0→7F2电偶极跃迁发射为主,发射波长为615 nm的红色光;具有反演中心对称性时,以5D0→7F1磁偶级跃迁发射为主,发射波长为587 nm的橙红色光.通过发射光谱图分析,在铕配合1和2中,Eu(Ⅲ)离子均处于非对称中心,发出明亮的红色光.

3 结 论
本文从设计具有优良发光性能的稀土铕配合物发光材料出发,选用两种具有较大共轭程度的β-二酮类第一配体:2-噻吩甲酰三氟丙酮(HTTA)和4,4,4-三氟-1-苯基-1,3-丁二酮(HBTA),分别与三苯基氧膦(TPPO)、六水合氯化铕(EuCl3·6H2O)反应,合成两种三价铕配合物.对于合成的铕配合物,通过氢核磁共振(1H NMR)、红外光谱(IR)、紫外光谱(UV)和X-射线单晶衍射对其结构进行表征.中心Eu(Ⅲ)离子分别与三个第一配体和两个第二配体配位,呈现八配位的多面棱镜形态.利用荧光光谱仪测试铕配合物1和2的荧光性质,发现在408 nm和398 nm波长的激发下,铕配合物1和2均在615 nm处发射出窄而尖锐的特征峰,其单色性好,是良好的红光发光材料.
猜你喜欢
有机氟工业(2022年3期)2022-09-29
浙江化工(2022年8期)2022-09-05
武汉工程大学学报(2022年4期)2022-08-26
四川大学学报(自然科学版)(2022年4期)2022-07-22
四川化工(2022年1期)2022-03-11
黄山学院学报(2021年5期)2021-11-06
化工设计通讯(2020年8期)2020-01-12
青岛大学学报(工程技术版)(2019年2期)2019-09-10
分析化学(2017年12期)2017-12-25
中学化学(2015年8期)2015-12-29
