
氮化碳光催化材料的掺杂改性研究进展
2020-06-24 01:30王福禄王建华李子强
精细石油化工 2020年3期
王福禄,王建华,李子强
(山东省医药工业设计院,山东 济南 250013)

在实际应用中,由于g-C3N4对可见光的利用效率较低,光生电子-空穴复合率高,低电导率和比表面积较小,导致其光催化活性较低[9-10]。其禁带宽度为2.7 eV,对应于约460 nm的临界吸收波长,使得光谱范围内的大部分可见光不能被吸收利用。因此,为了提高g-C3N4的光催化反应活性,人们从提升g-C3N4光学性能、比表面积、电荷分离效率效率等方面[11-20]对其进行了改性。
通过在半导体材料原子结构中引入杂原子可以改变其电子结构,从而对能带结构产生影响。杂原子的引入,特别是表面杂原子的引入可有效改变半导体材料的表面电子活性,可为催化反应提供更为优化的表面反应结构。因此,通过合适的掺杂改性,可以有效调控半导体材料的光吸收范围、电子传递能垒和表面面催化反应能垒,从而提升其整体的光催化性能。在g-C3N4框架结构中引入杂原子(金属和非金属掺杂、缺陷掺杂、自掺杂等)是提高材料光催化活性的有效措施。通过在g-C3N4中掺杂少量的金属或非金属元素,可以改变g-C3N4的电子结构和能带结构,达到拓宽g-C3N4的吸收波长,促进光生载流子的分离与传输能力,降低表面反应能垒等目的,从而有效提高g-C3N4的光催化性能。
由于近年来g-C3N4光催化材料的掺杂改性研究工作逐渐成为研究的热点,而到目前尚未有相应的报道对该领域做相应的总结和评述。因此本文从氮化碳的金属掺杂、非金属掺杂、层间掺杂、缺陷掺杂以及自掺杂等几个方面展开综述总结,并对该领域未来的方向做出展望。
1 掺杂对g-C3N4光催化材料能带结构的调控
金属元素掺杂的作用机理是掺杂的元素能够在价带上方产生施主能级或在导带下方产生受主能级,半导体吸收了光子的能量后,除了将电子从价带激发到导带外,也会将电子从施主能级激发到导带,或从价带激发到受主能级,从而使得g-C3N4的带隙变窄,吸光范围增大[21]。用于金属掺杂改性的元素范围十分广泛,如碱金属、过渡金属、稀土金属、贵金属以及非金属元素掺杂能在一定程度上提高g-C3N4的光催化产氢效率和环境污染物去除效率。
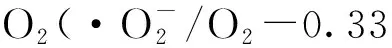

图1 金属锰离子通过配位路径修饰的g-C3N4骨架示意[24]
Ding等[23]发现,Fe3+,Mn3+,Co3+等过渡金属离子的引入并未改变g-C3N4的主体结构(图1)。与此同时,Fe和Cu掺杂的g-C3N4在H2O2存在的条件下能够有效促进苯羟基化生成苯酚,而Fe和Co掺杂的g-C3N4能促进苯乙烯的环氧化反应,这说明了过渡金属离子掺杂能有效通过改变g-C3N4的能带和电子性质,从而促进其光催化性能的提高。
同时,Fe作为一种常见的过渡金属元素,经常用于半导体材料的掺杂改性。Tonda等[25]以三聚氰胺和FeCl3为前驱体成功合成了Fe-g-C3N4纳米片,通过紫外-可见(UV-vis)漫反射光谱可以看出(图2),Fe-g-C3N4表现出明显的红移,增加了其在可见光范围内的吸收。


图2 Fe-g-C3N4纳米片材料的d紫外-可见分光反射光谱(a)和催化剂的光催化析氢循环稳定性(b)
非金属元素掺杂的作用机理不同于金属元素,它是通过掺杂元素与g-C3N4的价带发生杂化,从而提升价带顶来缩小禁带宽度,从而提高g-C3N4的光催化活性。非金属元素掺杂不会像金属元素掺杂那样引入复合中心,因而具有较好的光催化效果。
Man等[26]通过密度泛函理论计算(DFT)从理论上研究了P和S掺杂对g-C3N4的影响。他们分别通过原子取代和间隙掺杂两种形式模拟掺杂类型,接着利用第一性原理研究它们的掺杂形成能、电子性质以及可见光催化活性。发现,S原子优先取代位于边缘的N原子,而P原子优先进入位于g-C3N4平面内的间隙位置,非金属掺杂可降低带隙宽度,增强了g-C3N4的可见光吸收能力。
除了上述P掺杂g-C3N4材料之外,Zhu等[27]以三聚氰胺和磷酸为原料合成了分层堆积的P掺杂g-C3N4六角形管纳米结构,制备过程主要包括两个阶段:1)在磷酸辅助条件下水热反应(pH 1-3),三聚氰胺首先发生水解生成三聚氰酸,进而自组装形成六方棒状结构;2)同时在超分子前驱体表面吸附了磷酸分子,热处理后,P进入g-C3N4框架形成六角形管掺杂g-C3N4。通过P 2p XPS谱和31P核磁共振谱证实了g-C3N4中P的掺入(图3),类似地,P掺杂显著提高了材料的电导率和在可见光区的吸收。因此,该材料具有很高的产H2效率(67 mmol/h,0.1 g催化剂,I>420 nm),在420 nm处的表观量子效率(AQY)高达5.68%,明显高于大多数体相g-C3N4材料,具备良好的应用前景。

图3 超分子前驱体合成法制备的掺P管状g-C3N4(a)和P2p的高分辨XPS谱(b)以及掺杂P的g-C3N4的31P固态NMR谱(c)在(b)和(c)图中的插图为P在g-C3N4骨架中的替代掺杂结构。
Huang等[28]首先通过H2O2处理三聚氰胺得到氢键诱导的超分子聚集体,之后在550 ℃下煅烧得到了高度多孔网状结构的O掺杂g-C3N4材料(图4)。他们基于DFT的计算,发现:1)O掺杂优先发生在sp2杂化的N 原子位点,该位置的形成能最低;2)O掺杂能够有效增强光吸收。该材料的产氢效率分别是体相和三维多孔g-C3N4的6.1和3.1倍,在420 nm处的表观量子效率达到了7.8%。说明简单的前体预处理不仅可以控制框架结构,还可以引入有益的外来原子或单体,为高效光催化剂的设计和制备方法提供了一种新的策略。
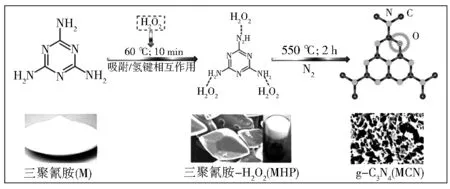
图4 氢键诱导超分子组装法制备同时具有多孔网络和O掺杂的g-C3N4的示意
Zhang等[29]采用双氰胺和碘化铵制备了碘掺杂g-C3N4光催化剂,通过理论计算(图5),I原子优先取代sp2键合的N原子。g-C3N4和I原子之间的亲密接触导致了π共轭芳香杂环的扩展,从EPR测试结果可以看出,I掺杂使得导带态密度增加了。因此,I掺杂能够有效增加g-C3N4的比表面积,延长光吸收以及促进电荷分离,从而提高了材料的产氢的效果。
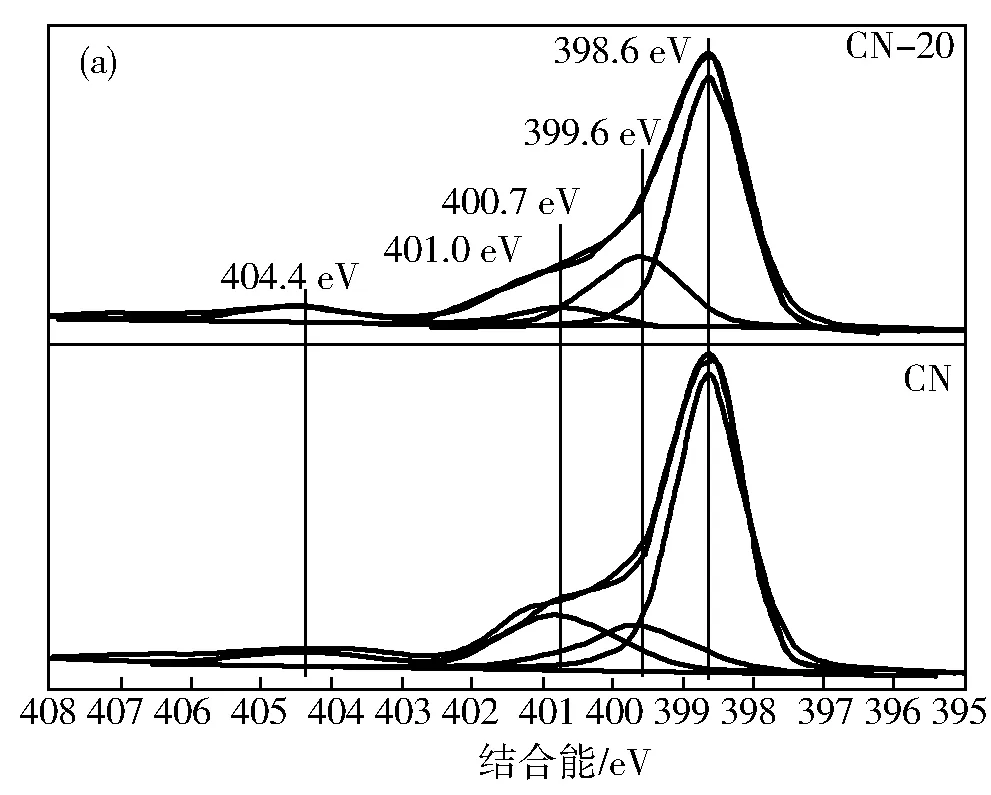

图5 CN和CN-l-20的XPS高分辨N 1s谱(a)和不同碘含量掺杂的CN试样的室温EPR谱(b)
通过在g-C3N4体相中引入缺陷掺杂位点也可以影响其禁带宽度。Niu等[30]通过控制缩聚反应温度,首次在g-C3N4的框架中成功引进了氮空位。他们发现:氮空位的存在能够有效窄化带隙,可促进g-C3N4在可见光区的吸收范围。典型的g-C3N4的N 1s主要由位于398.6 eV处的主峰和400.3 eV处的次峰组成(图6),分别对应于N sp2杂化的(N2c)键和N sp3杂化的(N3c)键,而形成氮空位后,N2c峰的强度明显变弱,说明在缩聚过程中形成的氮空位主要位于N2c处,这是由于N2c原子总量较多,且具有不饱和配位。Yu等[31]采用一种简单的碱辅助合成方法,即在g-C3N4的前驱体如尿素、三聚氰胺、硫脲中添加碱性化合物(如KOH),成功合成了含有N缺陷的g-C3Nx。得到的g-C3Nx具备可调节的能带结构(图7),从而能够更有效的捕获光能,在可见光下具有更好的产氢性能。更为重要的是,通过碱辅助方法可以方便的合成具有特定光学和光催化性能的g-C3N4材料,该法可能同样用来调整其他氮基半导体的能带结构,以改善其对太阳能的捕获和转换效率。

图6 反褶积高分辨率N 1s和c1s XPS光谱a—g-C3N4;b—氮缺陷g-C3N4

图7 纯g-C3N4和g-C3Nx的能带结构排列
2 掺杂对g-C3N4光催化材料光生载流子分离效率的优化
金属元素同样可用作掺杂元素来提升g-C3N4光催化材料的光生载流子分离效率。Chang等[32]制备了Pd修饰的介孔g-C3N4用于水体中双酚A(BPA)的降解。g-C3N4表面掺杂的Pd大多数以Pd0的形式存在,可以作为电子陷阱从而促进光生电子-空穴对的分离。以KI作为钾源合成的K掺杂g-C3N4光催化剂对苯酚和亚甲基蓝(MB)显示出较好的催化降解效果[33]。其中,K负载量为22%的22%K- g-C3N4试样光催化活性最高,对苯酚和MB的降解速率分别是纯g-C3N4的3.3和5.8倍。这同样是因为K掺杂有效的降低了g-C3N4的价带位置,从而促进了g-C3N4的氧化能力,提高了光生载流子的分离。
非金属元素掺杂的作用机理不同于金属元素,它是通过掺杂元素与g-C3N4的价带发生杂化,不会像金属元素掺杂那样引入复合中心,因而具有较好的促进光生载流子分离效率的作用。Zhou等[34]以六氯环三磷腈(Cl6N3P3)和盐酸胍作为前驱体,通过热聚合法制备了P掺杂g-C3N4。由于Cl6N3P3中的P-N杂环能够很好的与g-C3N4的基本结构3-s-三嗪环匹配,因此P原子能够进入到g-C3N4的间隙位置。P原子的引入显著改变了g-C3N4的电子性质,并且有效抑制了光生载流子的复合,因此显著提高了g-C3N4的光催化性能。Wang等[35]分别通过将硫脲和三聚氰胺在520 ℃下煅烧制备了S掺杂g-C3N4(TCN)和g-C3N4(MCN),通过理论计算来研究了TCN和MCN的局部态密度,发现TCN和MCN的带隙是相同的,但是TCN试样中存在其他杂质。因此,光生电子能很轻易地从杂质态跃迁到导带或从价带跃迁到杂质态,有利于光生载流子的分离。Zhou等[36]通过在前驱体尿素中添加少量柠檬酸成功合成了N掺杂g-C3N4。N掺杂除了能够增加g-C3N4材料的光吸收,抑制载流子复合以外,还能促进p-共轭系统的离域化,从而大大提高了其在可见光下分解水产H2的性能。Lin等[37]通过热聚合尿素和四苯硼钠(Ph4BNa)合成了B掺杂g-C3N4,其中,Ph4BNa不仅可以作为B的前体,也能作为尿素聚合改性剂。B原子和苯基离去基团可以在g-C3N4的碱性表面提供酸性基团,从而改变g-C3N4表面的化学性质。重要的是,这些酸性基团能够作为表面活性中心,这无疑更有利于光催化反应的进行。
对g-C3N4而言,除金属和非金属掺杂外,表面缺陷同样能够起到促进光生载流子分离的作用。Liang等[38]通过在NH3氛围下热处理体相g-C3N4(BGCN),得到了具有大量碳空位的多孔g-C3N4纳米片(HGCNs),其制备工艺如图8所示。NH3刻蚀不仅使BGCN剥落为含有大量孔隙的纳米片结构,比表面积高达196 m2/g,还提供了丰富的碳空位,改善了材料的化学结构。因此,与BGCN相比,HGCNs的产氢性能提高了约20倍,这主要得益于表面孔隙和碳空位促进了材料的光吸收和光生载流子分离效率。Guo等[39]以三聚氰胺为前驱体,通过焦磷酸盐辅助水热法合成了表面具有碳缺陷的P掺杂管状g-C3N4,作为对比,文中使用其他磷酸盐,如磷酸铵、次亚磷酸钠、亚磷酸钠,得到类似的P掺杂g-C3N4纳米管(图9),从而突出这种方法的通用性。其中,采用焦磷酸钠辅助水热合成的材料光催化活性最高,通过调节电子结构、表面缺陷、活性位点和结构特性来增强材料的可见光吸收和光生电子-空穴分离效率。

图8 HGCN纳米片制备工艺
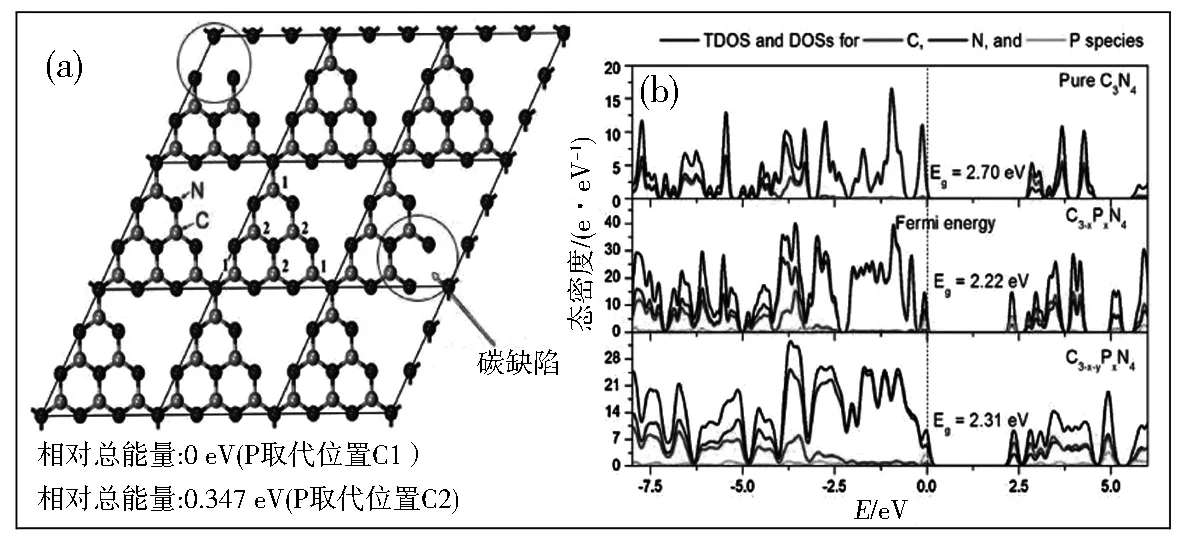
图9 基于g-C3N4理论模型,P取代C的不同位置时的相对总能量(a)和基于体相g-C3N4、P掺杂g-C3N4以及P掺杂g-C3N4的密度泛函理论(DFT)计算带结构(b)
3 掺杂对g-C3N4光催化材料光生载流子迁移过程的调控
光生载流子在半导体材料中的迁移率与其寿命成正相关,较高的迁移率以及优化的迁移路径可有效提高光催化材料的光催化性能。
金属掺杂可有效提高-C3N4光催化材料的电子迁移率。Zhang等[40]通过软化学法合成了Co-g-C3N4,Co改性后g-C3N4能够保持基本的石墨相半导体结构,但是材料光载流子迁移率及电子电导率明显提升。Co-g-C3N4的O2产生速率是纯g-C3N4的两倍。Co修饰能够从分子水平上提高Co-g-C3N4的光催化氧化能力,有着良好的应用前景。Xiong等[41]采用热聚合方法分别制备了K-C3N4和Na-C3N4光催化剂。研究发现:K嵌入到g-C3N4的层间结构中,而Na则倾向于存在于平面内(图10);K-C3N4对NO的光催化去除效果明显优于Na-C3N4试样,这是由于K原子进入g-C3N4的层间后,能拓展π共轭体系,有效减少电子局域态,从而促进光生载流子的在氮化碳层间的有效转移,提高了g-C3N4的光催化性能。
非金属掺杂也可以有效的调控氮化碳的光生载流子迁移率。Dong等[42]制备了C自掺杂g-C3N4,C原子取代了桥联的N原子,这可能会改变材料的电子和能带结构。理论计算结果表明C掺杂导致芳香环和C原子之间离域大π键的形成,从而增强了材料的导电性,有利于电子的转移。Liu等[43]通过共热解三聚氰胺和氯化铵制备了Cl插层掺杂g-C3N4,该试样具有多孔结构,有效增加了材料的比表面积,同时,由于建立了层间通道,从而促进了光生电荷的迁移。Niu等[44]通过H2处理制备了N缺陷修饰的g-C3N4,发现掺杂剂的空间分布是改变光催化剂电子结构的一个主要原因。在整个纳米颗粒均相掺杂适当的杂原子能够有效使带隙变窄,而表面掺杂则只能导致局域态的出现。对于非层状结构的材料来说,H2处理后通常是在表面形成阴离子空位,如图所示。而对于层状结构的材料来说,除表面缺陷外,层与层之间形成的缺陷则能够有效减少扩散路径。H2处理之后的g-C3N4带隙宽度减小到2.03 eV,层间的离子通道能够促进光生电荷的迁移,从而产生更多的·OH用于有机污染物的降解。
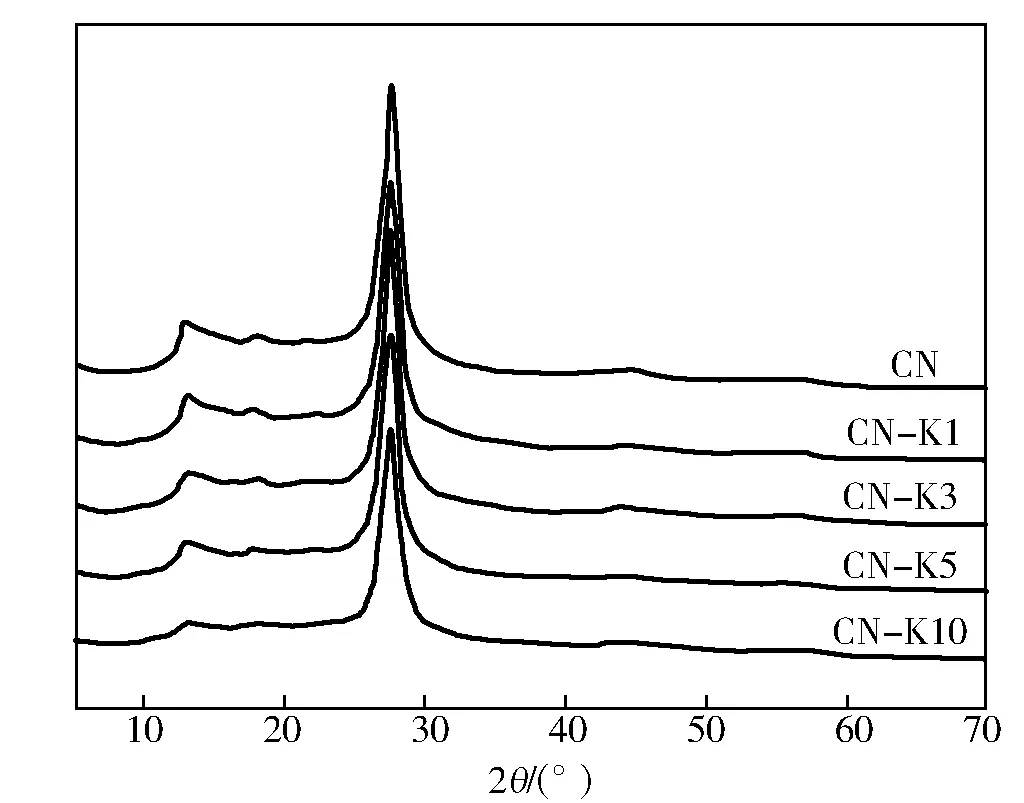

图10 不同掺钾量g-C3N4的x射线衍射曲线
4 总结及展望
在过去的几年中,人们致力于制备新型的掺杂改性g-C3N4复合材料,凭借其独特的结构特征和良好的光催化效果在环境污染治理和新能源的开发利用方面有着良好的应用前景。一般来说,对于改性g-C3N4复合材料光催化性能的评价标准分为3个方面:1)吸收区域向可见光区扩展;2)光生电子空穴的分离效率;3)光生载流子的迁移率。通过掺杂改性能够在分子水平上调节g-C3N4的能带结构,拓宽了材料的可见光吸收范围,提高了材料的氧化能力,同时有效抑制光生载流子的复合率。
在进行g-C3N4掺杂改性时,还应重视以下几方面:1)虽然掺杂元素的引入可以缩短其禁带宽度,但多以间接带隙的形式拓宽可见光区的吸收范围,使得可见光区的吸收强度不够;因此,需要开发形成直接带隙的掺杂元素和掺杂方式。2)对g-C3N4进行非金属掺杂时难以精确控制,易引入杂质元素,对结构的控制方法也比较单一,对光催化效果会造成一定影响,因此材料的合成方法需要进一步改进。3)针对g-C3N4的结构特征选择合适的掺杂方式。例如,g-C3N4具有类似石墨的层状结构,因此部分杂原子能够进入到层间结构中,形成离子传输通道,这种层间掺杂的方式可能是未来研究的新方向;氮化碳材料为层状堆叠的石墨相材料,其掺杂位点分为层间和层面内,层间掺杂可有效改善电子传递能力,面内掺杂可改变表面反应能垒,因而通过调控不同的掺杂位点,实现不同的功能调控。
猜你喜欢
科学之友(2022年11期)2022-11-03
工业水处理(2022年6期)2022-06-23
物理学报(2022年6期)2022-03-30
石油化工高等学校学报(2021年3期)2021-07-15
海洋通报(2020年5期)2021-01-14
物理学报(2020年16期)2020-08-29
中成药(2018年11期)2018-11-24
科技风(2018年9期)2018-05-14
上海农业学报(2017年3期)2017-04-10
原子能科学技术(2015年8期)2015-12-15
