
急性肾损伤miR-34a、GPS2表达变化及其对JNK通路的影响
2020-06-23 13:40陈伟伟王德选沈猛庄捷秋蔡晖
温州医科大学学报 2020年6期
陈伟伟,王德选,沈猛,庄捷秋,蔡晖
(温州医科大学附属第二医院育英儿童医院,浙江 温州 325027,1.肾内科;2.儿童肾内科)
急性肾损伤(acute kidney injury,AKI)是一种临床常见危重病,由多种病因造成肾功能在短期内急性减退[1]。由不同病因(如缺血、炎症、毒素等)导致的急性肾小管坏死(acute tubular necrosis,ATN)是最常见的AKI之一。国内外对常见ATN演变方向的干预研究还处于探索阶段[2]。因此在AKI后至纤维化的过程中找到新的治疗靶点显得尤为重要。
近年来,针对各类microRNAs在肾脏疾病中的表达谱研究很多,许多microRNAs被证明与AKI密切相关[3]。在AKI中,肾小管上皮细胞会过度表达miR-34a,参与调控肾纤维化和肾损伤[4]。G蛋白途径抑制因子2(G-protein pathway suppressor 2,GPS2)是G蛋白-MAPK途径的调节蛋白之一,它参与细胞骨架构建、DNA修复、炎症反应等体内多种生理病理过程。本课题组在动物实验中发现上述指标在肾缺血再灌注损伤中均出现显著的变化,但两者是否存在关联,有无存在通路蛋白的变化,以及干预后是否影响AKI后进入纤维化的进程尚无定论,为此本研究进行深入探索。
1 材料和方法
1.1 材料 健康成年雄性SD大鼠30只,12周龄,体质量250~300 g,无固定病原级,由北京维通利华实验动物技术有限公司(浙江嘉兴平湖基地)提供,饲养于温州医科大学实验动物中心,常规饲料和自来水按时无限制供应,动物许可证号:SCXK(浙)2018-0001。TNF-α、TGF-β ELISA试剂盒及引物由上海博蕴有限公司提供;HEK 293T细胞株由中华细胞库提供;miR-34a inhibitor、NC inhibitor、转染试剂均购自广州锐博公司;TB Green®Premix Ex TaqTMII(Tli RNaseH Plus)和PrimeScriptTMRT Master Mix(Perfect Real Time)购自大连Takara公司;GPS2等兔多克隆抗体来自美国Abcam公司;p-SPAK/JNK、 t-JNK、Beta-Actin等多克隆兔抗为美国Cell Signal Technology产品;二抗来自美国Jackson ImmunoResearch公司。其余试剂均为市售分析纯。
1.2 动物实验
1.2.1 动物分组与模型构建:30只SD大鼠喂养普通饲料适应1周后,随机分6组,为灌注对照0 h、24 h、 48 h组和灌注模型0 h、24 h、48 h组。大鼠注射2%戊巴比妥钠(40 mg/kg)麻醉后采用腹部切口暴露双侧肾蒂,模型组采用无损伤动脉夹夹闭肾蒂 25 min后开放灌注[3],缺血后肾脏呈现均一紫红色,移除动脉夹肾脏紫色消失,判断缺血再灌注成功。对照组开腹游离肾蒂不做夹闭,25 min后各组大鼠腹腔逐层缝合切口。手术过程大鼠置于恒温电热毯上维持37 ℃,等待麻醉药过后苏醒,观察大鼠反应存活情况,待确认存活后再将大鼠放回笼中。
1.2.2 标本采集:各组大鼠开腹前经股静脉取血2 mL,以测定造模前大鼠血清中TNF-α、TGF-β水平。造模成功后,按照灌注时间后将大鼠麻醉,获取血液和肾脏标本。取血后以3 000 r/min离心10 min,留取上层血清保存于-80 ℃冰箱,用于检测炎性及纤维化因子指标。离断下腔静脉,经左心室灌注0.9%氯化钠溶液,肾脏变白后取下肾脏,1/3肾脏组织放在4%组织细胞固定液固定,用于形态学观察;剩余肾脏组织保存至液氮中,用于后续mRNA和蛋白的检测。对照组同时间点标本采集同上。
1.2.3 血清TNF-α、TGF-β水平检测:依照ELISA试剂盒说明步骤进行检测。
1.2.4 肾组织形态学观察:取4%组织细胞固定液固定肾脏组织24 h后,脱水、石蜡包埋、切片(3 μm), 制成石蜡切片,HE染色,置于显微镜下(×400)观察肾小管上皮细胞形态。
1.3 细胞培养、转染与模型构建 人胚胎肾小管上皮细胞系(HEK 293T)培养在含10%胎牛血清的DMEM培养液中,常规添加100 μg/mL链霉素和100 U/mL 青霉素,培养箱条件为5% CO2,37 ℃,每2~3 d更换培养液。取对数生长期细胞于超净台下进行细胞分组,对照组(Control、NC inhibitor)和缺氧/复氧诱导AKI细胞模型组(Model、Model+miR-34a inhibitor)。将5×105个/mL密度细胞接种在24孔板中,正常培养,待细胞生长至50%~60%密度时,以100 nmol/L的终浓度在对应小组中转染NC inhibitor 及miR-34a inhibitor,转染48 h后,将模型组放在去血清和葡萄糖培养基中24 h,然后放入1% O2、94% N2、5% CO2缺氧条件下诱导6 h,再更换成完全培养基后放入5% CO2、95%空气条件下复氧30 min[5], 对照组置于5% CO2、95%空气条件下的培养箱中30 h 30 min,收集细胞进行下一步操作。
1.4 RT-qPCR检测miR-34a、GPS2 mRNA水平 用TRIzol裂解组织/细胞,氯仿、异丙醇萃取后离心,70%乙醇漂洗2次,20 min室温挥发、加DEPC水等步骤提取总RNA,测定RNAs浓度,依照反转录试剂盒说明反转录成cDNA。按照各组cDNA加1 μL+DEPC水 3.2 μL+前向引物0.4 μL+后向引物0.4 μL+TB Green 5 μL=10 μL体系加样,复孔3个,热循环进行PCR反应。条件如下:95 ℃初始变性30 s,40个循环(95 ℃下5 s,60 ℃下30 s),融解阶段(95 ℃ 下15 s,60 ℃下30 s,95 ℃下15 s),置于罗氏PCR仪上检测。以上实验重复进行3次。选择U6和GAPDH作为内参,按照2-ΔΔCT计算各基因相对表达量。引物序列:U6-qPCR-F:CTCGCTTCGGCAGCACA,U6-qPCR-R: AACGCTTCACGAATTTGCGT;miR-34a F-5P-qPCR-F:ACA CTCCAGCTGGGTGGCAGTGTCTTAGCT,miR-34a R-5PqPCR-R:CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGA CAACCAG;Rat-GAPDH-qPCR-F:GCAAGTTCAACGGCACAG, Rat-GAPDH-qPCR-R:GCCAGTAGACTCCACGACAT;Rat-GPS2-qPCR-F:AGTACCTCTGAGCTCCCACTT,Rat-GPS2- qPCR-R:CATGGCGTTGGAAAGCTTGG;Human-GAPDHqPCR-F:AAGAAGGTGGTGAAGCAGG,Human-GAPDH-qPCRR:GTCAAAGGTGGAGGAGTGG;Human-GPS2-qPCR-F: AGTGACCTGACCACCCTAACA,Human-GPS2-qPCR-R:CCT GGGCGATTGTGTCCTC。
1.5 Western blot检测GPS2、p-JNK蛋白表达 用RIPA裂解液裂解肾脏组织和细胞,冰上静置30 min 后超声裂解,4 ℃下12 000 r/min离心20 min留取上清液。配平各组蛋白浓度,各加入预热的5× Loading Buffer。37 ℃水浴30 min用于检测GPS2,actin,100 ℃煮沸5 min用于p-JNK蛋白检测。每孔加入30 μg蛋白样品进行10%聚丙烯酰胺凝胶蛋白电泳,浓缩胶电压80 mV,分离胶电压120 mV。
冰浴下100 V恒压半干式转膜,转膜时间根据蛋白分子量设置60 min到150 min不等。依次孵育一抗(多克隆兔抗GPS2抗体1:1 000;多克隆兔抗JNK抗体1:1 000;多克隆兔抗Beta-Actin抗体1:1 000,4 ℃摇床孵育过夜)、二抗(HRP标记山羊抗兔二抗1:7 000,水平摇床上室温孵育60 min),ECL显色。使用凝胶图像处理系统对目的条带进行量化分析。以上所有实验重复4次。
1.6 统计学处理方法 采用SPSS22.0、ImageJ和GraphPad软件进行数据分析、制图。计量资料以 ±s表示,多组间比较采用单因素方差分析,组间两两比较方差齐性采用LSD检验,方差不齐采用Dunnett T3检验。P<0.05为差异有统计学意义。
2 结果
2.1 大鼠缺血再灌注不同时间段肾小管细胞形态结构改变 对照组(0 h、24 h、48 h)肾小管上皮细胞结构正常,肾小球及肾小管未见明显异常改变(见图1A、C、E);与同时段对照组相比,模型组发生不同程度的损伤,0 h时肾小管上皮细胞肿胀,管腔扩张,刷状缘消失;24 h时肾小管结构排列紊乱、松散,间质部分狭窄和炎性细胞浸润;48 h时除上述变化外,还可见到蛋白管型增多(见图1B、D、F)。

图1 相差显微镜观察缺血再灌注不同时间肾小管细胞形态结构改变(× 400,箭头示肾小管细胞空泡化、刷状缘消失、炎性细胞浸润及蛋白管型增加)
2.2 大鼠缺血再灌注不同时间段炎症及纤维化指标变化 与对照组(0 h、24 h、48 h)比较,模型组同时段TNF-α、TGF-β表达水平显著升高(均P<0.01);与造模前相比,模型组造模后各时段TNF-α、TGF-β表达水平明显升高,差异有统计学意义(均P<0.01),TNF-α、TGF-β表达在24 h达到最高峰(P<0.01)。见表1。
2.3 大鼠缺血再灌注不同时间段miR-34a、GPS2 mRNA表达变化 PCR结果显示:对照组0 h和模型组0 h时的miR-34a mRNA水平和GPS2 mRNA水平差异均无统计学意义(P>0.05)。与对照组24 h和 48 h比较,模型组相应时段miR-34a mRNA水平明显升高(均P<0.01),GPS2 mRNA水平显著下降(均 <0.01),随着灌注时间延长,miR-34a mRNA水平呈增加趋势,而GPS2 mRNA水平呈下降趋势(均P<0.01),见图2。
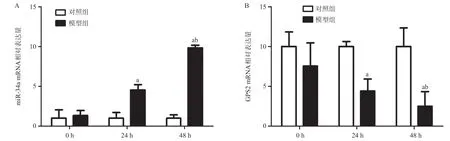
图2 大鼠缺血再灌注不同时段miR-34a、GPS2 mRNA水平变化

表1 大鼠缺血再灌注不同时间段TNF-α和TGF-β水平变化(每组n=5,±s,ng/L)
2.4 大鼠缺血再灌注不同时间段GPS2、p-JNK蛋白表达变化 Western blot显示:与各自对照组比较,模型组GPS2蛋白表达下降,p-JNK蛋白表达升高,差异有统计学意义(均P<0.05);灌注时间在48 h时,GPS2表达下降及p-JNK表达升高最显著(P<0.01),见表2。
2.5 HEK 293T应用miR-34a inhibitor后miR-34a、GPS2 mRNA水平 PCR结果显示:对照组(Control)与阴性对照组(NC inhibitor)之间miR-34a、GPS2 mRNA水平差异无统计学意义(P>0.05);与Control 组和NC inhibitor组相比,Model组的miR-34a上调,GPS2 mRNA水平下降,差异有统计学意义(均P<0.05);与Model组比,沉默miR-34a基因后,GPS2 mRNA水平上调,差异有统计学意义(均P<0.05),见图3。
2.6 HEK 293T应用miR-34a inhibitor后GPS2、p-JNK蛋白表达变化 Western blot显示:对照组(Control)与阴性对照组(NC inhibitor)之间GPS2蛋白表达及p-JNK蛋白表达无明显变化(P>0.05);与Control组和NC inhibitor组比较,Model组的GPS2蛋白表达减少,而p-JNK蛋白表达增加,差异有统计学意义(均P<0.05);与Model组比,沉默miR-34a基因后,GPS2蛋白表达增加,p-JNK蛋白表达减少,差异有统计学意义(均P<0.05),见表3。

表2 大鼠缺血再灌注不同时间段GPS2、p-JNK蛋白相对表达量变化(每组n=5,±s)
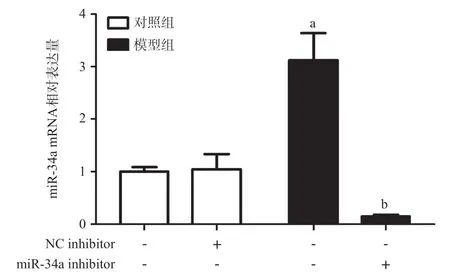
图3 HEK 293T应用miR-34a inhibitor后miR-34a、GPS2 mRNA水平变化
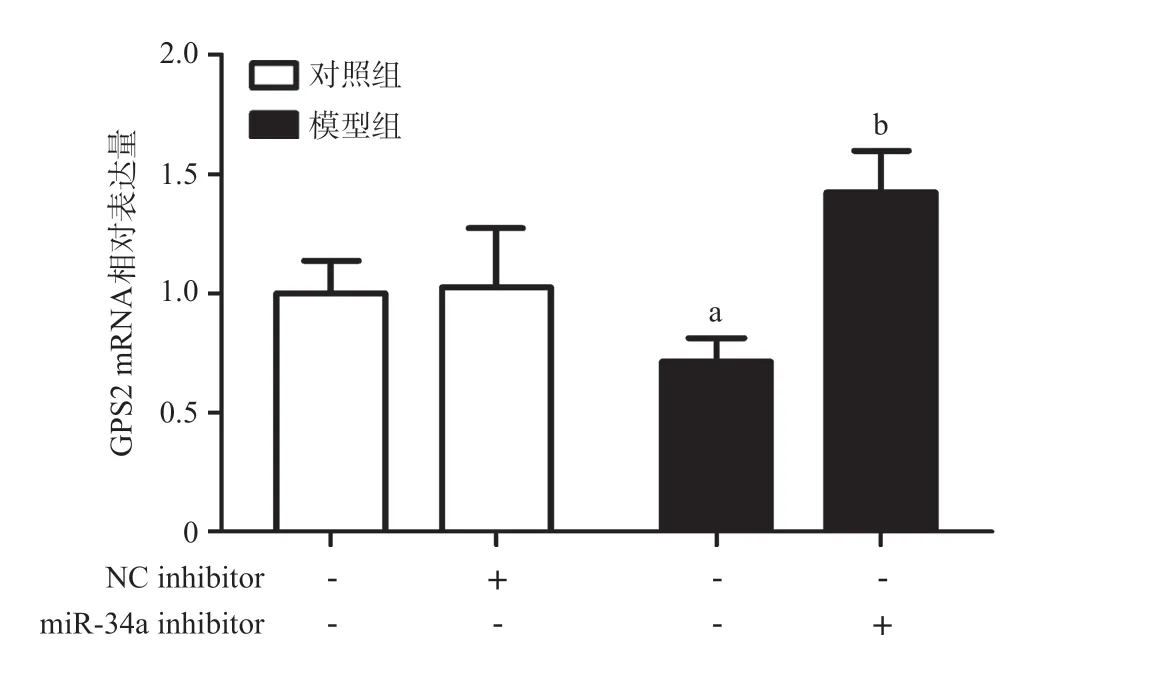
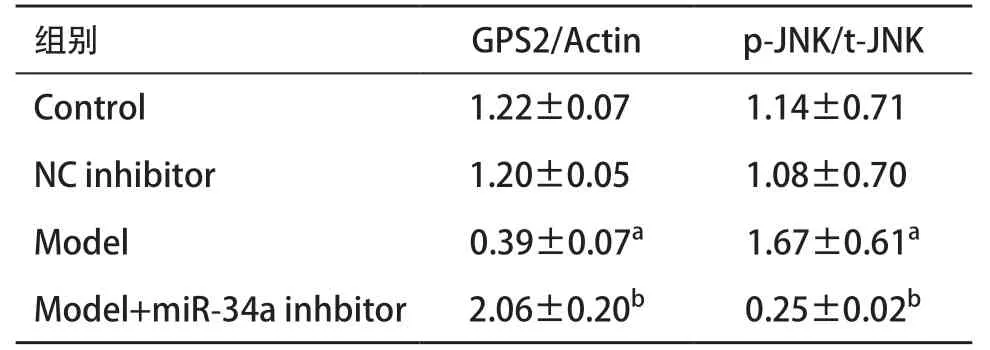
表3 HEK 293T应用miR-34a inhibitor后GPS2、p-JNK蛋白表达变化(每组n=3,±s)
3 讨论
临床上AKI的治疗主要以消除致病因素、对症治疗为主,但被动等待肾小管细胞自行修复,其中部分患者可能会出现肾脏不完全修复、进展性纤维化等情况,从而造成不同程度的肾功能异常[6-7],研究AKI进展至肾脏纤维化的可能机制并加以干预,对防治慢性肾脏病具有重要的意义。
MicroRNAs是一类长度约18~25个核苷酸的非编码小RNA分子,它们主要通过诱导mRNA裂解调节转录后的靶基因表达或干扰靶蛋白翻译水平起作 用[8]。本课题组在动物实验中发现,miR-34a在缺血再灌注引起AKI后出现GPS2表达下调的现象。miR-34a是一种多功能调节因子,参与糖尿病肾病[9]或疾病纤维化病理的过程[10],调节细胞增殖、凋亡和生长。GPS2在人体中许多组织都有表达,与许多蛋白相互作用,涉及细胞炎症、细胞周期调节等多种生理病理活动[11-13]。文献报道,miR-34a在AKI中过表达,可以靶向抑制SIRT1蛋白表达[14-15],但miR-34a是否还可能调节其他靶向蛋白,目前国内外尚少研究。本课题组通过细胞实验发现,miR-34a经缺氧再复氧的损伤后表达上调,GPS2的mRNA水平和蛋白表达均显著下降;沉默miR-34a基因,GPS2的mRNA水平和蛋白表达均明显增加。结合动物和细胞实验,我们推测miR-34a在AKI中可能下调GPS2表达。
损伤涉及炎症反应,干预不及时很快进入纤维化层面。在严重缺血、毒素和持续性炎症反应下,肾小管上皮细胞出现变性、脱落、坏死、凋亡,IL-1β、IL-8等因子参与炎症细胞的趋化与活化,使得炎症反应加重[2];损伤的肾小管上皮细胞激活蛋白激酶ATM[16],启动P53/P21通路,进而激活细胞内JNK信号通路,促进纤维化生长因子(TGF-β1和CTGF)的基因转录以及蛋白合成与分泌,作用于肾间质的成纤维细胞,促进其增殖和分化,导致肾间质纤维化[17]。本研究HE染色结果显示,肾小管细胞在缺血再灌注后发生了不同程度的损伤,可见上皮细胞肿胀、空泡化、管腔扩张及刷状缘消失等病理变化,表明大鼠肾脏缺血再灌注后出现明显的肾小管形态学损伤,且随着再灌注时间延长,其损伤加重。在缺血再灌注损伤后24 h和48 h,TNF-α、TGF-β表达水平明显升高,说明存在从AKI向纤维化转化的可能性。这一结果与文献报道[18-19]相一致,并提示造模成功。JNK信号通路是MAPK信号通路途径的分支,参与机体炎症及纤维化。文献报道GPS2能够特异性抑制JNK信号通路,而对其余的MAPK途径如P38通路则没有抑制作用[20]。本研究发现,miR-34a在缺血再灌注引起的AKI后,GPS2蛋白表达下降,p-JNK蛋白表达增加;HEK 293T细胞经过缺氧/复氧模拟AKI后,GPS2的mRNA水平和蛋白表达下降,而p-JNK蛋白表达增加;沉默miR-34a基因后,GPS2的mRNA水平和蛋白表达明显增加,p-JNK蛋白表达显著下降,这一结果和CARDAMONE等[21]发现GPS2通过抑制JNK通路在抗炎反应中起着重要的作用相符合。
综上所述,我们的动物和细胞实验初步证明,miR-34a可能通过下调GPS2蛋白,间接激活JNK信号通路进而影响受损肾小管上皮细胞DNA修复,促进AKI发生和发展。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中老年保健(2022年3期)2022-08-24
中老年保健(2022年3期)2022-08-24
中国现代医生(2022年21期)2022-08-22
传染病信息(2022年3期)2022-07-15
老年医学研究(2021年5期)2022-01-19
现代临床医学(2021年6期)2021-11-20
健康之家(2020年15期)2020-05-08
中国医学创新(2019年9期)2019-08-19
滨州医学院学报(2016年2期)2016-05-27
