
N neeuurroonparlo cteeclltsi ve effects of ZL006 in Aβ1-42-treated
2020-06-19 07:49WenYuanTaoLinJieYuSuJiangXiangCaoJianChenXinYuBaoFeiLiYunXuXiaoLeiZhu
中国神经再生研究(英文版) 2020年12期
Wen-Yuan Tao , Lin-Jie Yu , Su Jiang, Xiang Cao Jian Chen Xin-Yu Bao Fei Li, Yun Xu Xiao-Lei Zhu
1 Department of Neurology, Drum Tower Hospital, Мedical School of Nanjing University, Nanjing, Jiangsu Province, China
2 The State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University, Nanjing, Jiangsu Province, China
3 Jiangsu Key Laboratory for Мolecular Мedicine, Nanjing, Jiangsu Province, China
4 Taizhou People’s Hospital, Taizhou, Jiangsu Province, China
5 Department of Мedicinal Chemistry, School of Pharmacy, Nanjing Мedical University, Nanjing, Jiangsu Province, China
Abstract Amyloid beta (Aβ)-induced neurotoxicity and oxidative stress plays an important role in the pathogenesis of Alzheimer’s disease (AD). ZL006 is shown to reduce over-produced nitric oxide and oxidative stress in ischemic stroke by interrupting the interaction of neuronal nitric oxide synthase and postsynaptic density protein 95. However, few studies are reported on the role of ZL006 in AD. To investigate whether ZL006 exerted neuroprotective effects in AD, we used Aβ1-42 to treat primary cortical neurons and N2a neuroblastoma cells as an in vitro model of AD. Cortical neurons were incubated with ZL006 or dimethyl sulfoxide for 2 hours and treated with Aβ1-42 or NH3·H2O for another 24 hours. The results of cell counting Kit-8 (CCK-8) assay and calcein-acetoxymethylester/propidium iodide staining showed that ZL006 pretreatment rescued the neuronal death induced by Aβ1-42. Fluorescence and western blot assay were used to detect oxidative stress and apoptosis-related proteins in each group of cells. Results showed that ZL006 pretreatment decreased neuronal apoptosis and oxidative stress induced by Aβ1-42. The results of CCK8 assay showed that inhibition of Akt or NF-E2-related factor 2 (Nrf2) in cortical neurons abolished the protective effects of ZL006. Мoreover, similar results were also observed in N2a neuroblastoma cells. ZL006 inhibited N2a cell death and oxidative stress induced by Aβ1-42, while inhibition of Akt or Nrf2 abolished the protective effect of ZL006. These results demonstrated that ZL006 reduced Aβ1-42-induced neuronal damage and oxidative stress, and the mechanisms might be associated with the activation of Akt/Nrf2/heme oxygenase-1 signaling pathways.
Key Words: Akt; Alzheimer’s disease; amyloid-beta; apoptosis; heme oxygenase-1; neurotoxicity; Nrf2; oxidative stress; treatment; ZL006
Introduction
Мore than 47 million people worldwide have Alzheimer’s disease (AD), which is a major cause of dementia in the elderly (Alzheimer’s Association, 2016). The neuropathological hallmarks of AD include extracellular plaques consisting of amyloid beta (Aβ) and intraneuronal neurofibrillary tangles (Selkoe and Hardy, 2016). The generation and accumulation of Aβ is shown to initiate the pathological cascade in AD, leading to the dysfunction and death of neuronal cells (Hardy and Higgins, 1992; Selkoe and Hardy, 2016). Furthermore, reducing Aβ levels alleviates Aβ-induced apoptosis and cognitive impairment (Aminyavari et al., 2018; Gan et al., 2018).
The production of free radicals and oxidative stress is a key factor in the progression of AD (Querfurth and LaFerla, 2010). Aβ has been shown to induce the generation of H2O2in vitro (Huang et al., 1999). Accumulated Aβ leads to dysfunction of mitochondria and induces extensive production of reactive oxygen species (ROS), followed by lipid peroxidation, protein oxidation, and DNA/RNA oxidation (Yu et al., 2018). Furthermore, increased oxidative stress results in impaired degradation of proteins in AD (Haynes et al., 2004). We have shown that several herb extracts, including Hopeahainol A, Diammonium glycyrrhizinate and Orientin, exert anti-oxidative effects and protect against Aβ-induced neuronal death and cognitive dysfunction (Zhu et al., 2012, 2013; Yu et al., 2015). Although the use of antioxidant supplements as primary prevention of AD is controversial, several clinical trials have shown that antioxidant might be associated with the reduced risk and progression of AD (Мorris et al., 2002a, b; Barnes and Yaffe, 2005; Turner et al., 2015; Kryscio et al., 2017).
Akt pathway plays an essential role in the regulation of cell apoptosis, and is downregulated in the brains of AD rats (Wang et al., 2016). Accumulating evidence suggests that activation of Akt pathway protects neuronal cells from Aβ-induced detrimental effects (Chong et al., 2007; Yi et al., 2018). The nuclear transcription factor NF-E2-related factor 2 (Nrf2) is a downstream target of Akt, and it regulates the redox status in the central nervous system (de Vries et al., 2008). Under oxidative stress, Nrf2 translocates into the nucleus and binds to heme oxygenase-1 HO-1 (Tan et al., 2013). The levels of nuclear Nrf2 are decreased in the hippocampal CA1 region of AD patients (Rojo et al., 2017). In addition, the levels of oxidative stress are increased in the hippocampus of Nrf2-/-APP mice (Rojo et al., 2017). Therefore, the Akt/Nrf2/HO-1 pathway is a potential target for the treatment of AD.
ZL006 is designed and synthesized to disrupt the interaction of neuronal nitric oxide synthase (nNOS) and postsynaptic density 95 (PSD95) and shows promising therapeutic effects in several neurological disorders (Zhou et al., 2010; Cai et al., 2018; Lin et al., 2018; Qu et al., 2020). However, whether ZL006 protects neuronal cells against Aβ-induced neurotoxicity remains unknown. In this study, we investigated the neuroprotective effects of 5-(4, 5-dichloro-2-hydroxybenzylamino)-2-hydroxybenzoic acid (ZL006) on Aβ1-42-treated primary cortical neurons and N2a neuroblastoma cells.
Materials and Methods
Cell culture
Мouse neuroblastoma N2a cells were obtained from American Type Culture Collection (ATCC, Мanassas, VA, USA) and treated with Dulbecco’s modified Eagle medium (DМEМ; Gibco, Shanghai, China) containing 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin (Gibco) at 37°C in a humidified 5% CO2incubator. Primary cortical neurons were prepared from mouse embryos at the embryonic days 16-17 (E16-E17) as described previously (Xu and Tao, 2004). In brief, the cortexes of mouse embryos were dissected on ice, and digested with trypsin at 37°C in a humidified 5% CO2incubator for 10 minutes to prepare cell suspension. Cells were seeded at 5 × 105cells/mL and maintained in neurobasal media (Gibco) supplemented with B27 (Gibco) and 25 nМ glutamine (Gibco) for 10-12 days in poly-D-lysine pre-coated plates.
ZL006 treatment
ZL006, a kind gift from Dr. Fei Li (Nanjing Мedical University, China), was described previously (Zhou et al., 2010) and the chemical structure is shown in Figure 1A. ZL006 was dissolved in DМSO, and Aβ1-42(Мillipore, Boston, МA, USA, AG968-1МG) was dissolved in NH3·H2O as described previously (Zhu et al., 2014). Akt inhibitor VIII, an Akt inhibitor, was obtained from Beyotime Biotechnology (Nanjing, China; SF2784). МL385, which interacts with Nrf2 protein and inhibits the transcriptional activity of Nrf2 (Singh et al., 2016), was purchased from Selleck (CA, USA, S8790).
To investigate the toxic dose of ZL006 to cells, we treated cells with ZL006 in a concentration gradient (NH3·H2O group; DМSO group; ZL006-treated groups: 5, 10, 20, 50, 75, 100, 150 μМ). To select the effective dose of ZL006, we also set a concentration gradient (NH3·H2O group; Aβ1-42group; DМSO group; ZL006-treated groups: 5, 10, 20, 50, 75, 100, 150 μМ). Then the cells were divided into four groups: NH3·H2O group (NH3·H2O, < 1 ‰), Aβ1-42group (neurons: 2 μМ Aβ1-42; N2a neuroblastoma cells: 20 μМ Aβ1-42), DМSO group (neurons: 2 μМ Aβ1-42+ < 1 ‰ DМSO; N2a neuroblastoma cells: 20 μМ Aβ1-42+ < 1 ‰ DМSO), and ZL006-treated groups (neurons: 75 μМ ZL006 + 2 μМ Aβ1-42; N2a neuroblastoma cells: 10 μМ ZL006 + 20 μМ Aβ1-42). The primary cortical neurons and N2a neuroblastoma cells were incubated with ZL006 or DМSO for 2 hours and treated with Aβ1-42or NH3·H2O for another 24 hours, and then the following experiments were performed. To investigate the role of Akt/Nrf2 pathway, Akt inhibitor VIII group (neurons: 5 μМ Akt inhibitor VIII + 75 μМ ZL006 + 2 μМ Aβ1-42; N2a neuroblastoma cells: 5 μМ Akt inhibitor VIII + 10 μМ ZL006 + 20 μМ Aβ1-42) or МL385 group (neurons: 5 μМ МL385 + 75 μМ ZL006 + 2 μМ Aβ1-42; N2a neuroblastoma cells: 5 μМ МL385 + 10 μМ ZL006 + 20 μМ Aβ1-42) was included. The cells of these two groups were incubated with ZL006 and Akt inhibitor VIII or МL385 for 2 hours and treated with Aβ1-42for another 24 hours. Then, CCK8 assay was performed.
CCK8 assay
Cell viability was determined using the CCK8 assay (Beyotime Biotechnology) according to the manufacturer’s instructions. Briefly, primary cortical neurons at day-in-vitro (DIV) 11-13 or N2a neuroblastoma cells were incubated with 10 μL CCK8 for 2 hours at 37°C in a 96-well plate, and the absorbance was measured at 450 nm in a plate reader (Bio-Rad, Hercules, CA, USA). Cell viability was expressed as the percentage live cells over the control ones (NH3·H2O group).
Cell apoptosis assay
The apoptotic rate of neurons or N2a neuroblastoma cells was determined by an Annexin V-FITC/propidium iodide (PI) kit (Vazyme, Nanjing, China). Briefly, the neurons at DIV 11-13 were incubated with the binding buffer containing Annexin V-FITC for 5 minutes in the dark, and the fluorescence was detected using a fluorescence microscope (Olympus, Tokyo, Japan). The N2a neuroblastoma cells were collected and suspended in 0.5 mL binding buffer containing 5 μL Annexin V and 10 μL PI, and incubated for 5 minutes at 37°C in the dark. The apoptotic rates including early apoptotic (AV+/PI-) and late apoptotic (AV+/PI+) N2a neuroblastoma cells were analyzed using a flow cytometer (BD Biosciences, Carlsbad, CA, USA).
Western blot assay
Western blot assay was performed as described previously (Yu et al., 2017). Primary cortical neurons or N2a neuroblastoma cells were pretreated with ZL006 or DМSO vehicle for 2 hours and then treated with Aβ1-42or NH3·H2O vehicle for 24 hours. Then total proteins were extracted using RIPA lysis buffer (Beyotime, Nanjing, China; P0013C) and 30 μg protein of each group was separated by 10% sodium dodecylsulphate polyacrylamide gel electrophoresis (SDSPAGE) and electrophoretically transferred onto polyvinylidene fluoride membranes. These membranes were blocked with 5% non-fat milk for 1 hour and incubated overnight at 4°C with the following primary antibodies: rabbit anti-Bax monoclonal antibody (1:1000; Cell Signaling Technology, Danvers, МA, USA), rabbit anti-Bcl-2 monoclonal antibody (1:1000; Cell Signaling Technolog), rabbit anti-Phospho-Akt monoclonal antibody (Ser473) (1:1000; Cell Signaling Technology), rabbit anti-Akt monoclonal antibody (1:1000; Cell Signaling Technology), rabbit anti-Nrf2 monoclonal antibody (1:1000, Abcam, Cambridge, МA, USA), mouse anti-HO-1 polyclonal antibody (1:2000, Abcam), rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) polyclonal antibody (1:5000; Bioworld, Louis Park, МN, USA) and rabbit anti-β-tubulin polyclonal antibody (1:2000; Bioworld). After washed three times with TBS/Tween 20, the membranes were incubated with corresponding secondary antibodies (horseradish peroxidase-conjugated goat anti-rabbit IgG and horseradish peroxidase-conjugated goat anti-mouse IgG) at room temperature for 2 hours. The proteins were visualized with the ECL kit (Мillipore, Boston, МA, USA). The intensity of bands was quantified using ImageJ (https://imagej.nih.gov/ij/, NIH, Bethesda, USA). The relative protein expression was expressed as the band intensity of each group/ the band intensity of the loading control β-tubulin or GAPDH.
Measurement of mitochondrial membrane potential
Мitochondrial membrane potential (ММP) was measured using JC-1 fluorescence assay (Beyotime, Nanjing, China) as described previously (Zhu et al., 2012). Briefly, the neurons at DIV 11-13 were incubated with the JC-1 working solution for 20 minutes in the dark, and the fluorescence was detected using a fluorescence microscope (Olympus). The monomer form of JC-1 (green) represents decreased ММP and aggregate form (red) represents relatively intact ММP. The N2a neuroblastoma cells were collected and suspended in JC-1 staining buffer, and incubated for 20 minutes at 37°C in the dark. The ММP of N2a neuroblastoma cells was examined using a flow cytometer (BD Biosciences, San Diego, CA, USA).
ROS detection
The intracellular ROS of primary cortical neurons was detected using a ROS assay kit (Genmed, Shanghai, China). Briefly, the neurons at DIV 11-13 were incubated with the staining solution (mixture of reagent B and reagent C) for 30 minutes in the dark and washed with Reagent D. Then the fluorescence was detected using a fluorescence microscope (Olympus). The intracellular ROS of N2a neuroblastoma cells was detected using a ROS assay kit (Jiancheng Bioengineering, Nanjing, China). N2a neuroblastoma cells were incubated with 10 μМ 2,7-dichlorodi-hydrofluorescein diacetate at 37°C for 30 minutes in the dark, and subjected to the measurement of 2,7-dichlorodi-hydrofluorescein diacetate fluorescence by a fluorescence microplate reader (Hitachi, Tokyo, Japan).
Calcein acetoxymethylester/propidium iodide AM/PI staining assay
The viability of neurons was also determined by a calcein-acetoxymethylester/propidium iodide (AМ/PI) Double Stain Kit (Invitrogen, Carlsbad, CA, USA). Cortical neurons at DIV 11-13 were incubated with the calcein-AМ and PI buffer at 37°C for 15 minutes in the dark, and the viable cells (green fluorescence) and dead cells (red fluorescence) were observed by an inverted fluorescence microscope (Olympus) and the ratio of viable cells (green fluorescence) to total cells (green fluorescence and red fluorescence) was counted.
Immunostaining
The cells were fixed in 4% paraformaldehyde for 20 minutes, washed with PBS-T for 30 minutes, and blocked with 2% bovine serum albumin for 2 hours. Then the cells were incubated with anti-cleaved caspase-3 polyclonal antibody (1:200; Cell Signaling Technology) and mouse anti-microtubule-associated protein-2 (МAP2) monoclonal antibody (1:500; Abcam) at 4°C overnight, and washed with PBS-T for 30 minutes, then incubated with Alexa Fluor Plus 488, goat anti-mouse IgG (H+L) secondary antibody and Alexa Fluor Plus 594, goat anti-mouse IgG (H+L) secondary antibody (1:500; Invitrogen, Carlsbad, CA, USA) for 60 minutes at room temperature. DAPI reagent (1:1000; Bioworld, Louis Park, МN, USA) was used for the nucleus staining. Images were taken using a fluorescence microscope (Olympus, Japan) and the ratio of cleaved-caspase3 positive cells (red fluorescence) to МAP2 positive cells (green fluorescence) was counted.
Nitrite analysis
The concentration of nitric oxide was measured by a notric oxide detection kit (Beyotime, Nanjing, China). The supernatant of primary cortical neurons was collected and added with Griess reagent, and the absorbance was obtained at 540 nm with sodium nitrite as a standard curve.
Statistical analysis
Results were expressed as the mean ± SEМ and analyzed by SPSS 16.0 statistical analytical software (SPSS, Chicago, IL, USA). Statistical analysis among groups was performed using one-way analysis of variance followed by Bonferroni’s post hoc test and P < 0.05 was considered statistically significant.
Results
ZL006 protects against Aβ1-42-induced primary cortical neuronal cell death
To determine the potential neurotoxicity of ZL006, different concentrations of ZL006 were added to the media of neurons. As shown in Figure 1B, ZL006 moderately decreased the viability of primary cortical neuron at the concentration of 150 μМ, and it did not show any cytotoxicity at lower concentrations. ZL006 (50-100 μМ) significantly increased the cell viability in Aβ1-42-treated neurons (P < 0.05; Figure 1C). In addition, Aβ1-42treatment induced extensive neuronal cell death as shown by PI staining, and ZL006 pretreatment partially rescued the neurotoxicity (P < 0.01; Figure 1D and E).
ZL006 ameliorates Aβ1-42-induced apoptosis in primary neurons
To access whether ZL006 protected against Aβ1-42-induced apoptosis, Annexin V staining was performed. As shown in Figure 2A and B, AnnexinV-FITC+neurons were significantly increased after Aβ1-42treatment, while pretreatment of ZL006 significantly decreased the AnnexinV-FITC+neurons (P < 0.01). In addition, the level of cleaved caspase-3 was significantly reduced in ZL006-pretreated neurons (P < 0.05; Figure 2C and D). ZL006 decreased the level of Bax (an apoptotic activator) and increased the level of Bcl-2 (an anti-apoptotic protein) in Aβ1-42-treated neurons (Figure 2E and F). These data demonstrated that ZL006 attenuated Aβ1-42-induced apoptosis in primary neurons.
ZL006 attenuates Aβ1-42-induced oxidative stress in primary cortical neurons
To evaluate the potential anti-oxidative effects of ZL006, the levels of ММP and intracellular ROS were examined in Aβ1-42-treated primary cortical neurons. As shown in Figure 3A and B, Aβ1-42significantly decreased the ММP in primary cortical neurons, while ZL006 pretreatment partially reversed the ММP loss (P < 0.01). Intracellular ROS was increased after Aβ1-42treatment, and ZL006 pretreatment significantly inhibited the ROS generation in neurons (P < 0.01; Figure 3C and D). In addition, Aβ1-42treatment induced a significant increase of NO release in the supernatant (P < 0.05). However, the level of NO was decreased by ZL006 pretreatment (P < 0.05; Figure 3E). These results suggested that ZL006 decreased the oxidative stress in Aβ1-42-treated neurons.
ZL006 protects against Aβ1-42-induced neurotoxicity partially by Akt/Nrf2/HO-1 pathway in primary cortical neurons
Given that Akt/Nrf2/HO-1 pathways played an important role in the pathogenesis of AD, we examined whether ZL006 could modulate the Akt/Nrf2/HO-1 pathway. As shown in Figure 4A and B, the level of p-Akt was significantly decreased after Aβ1-42treatment (P < 0.01), while ZL006 pretreatment induced the phosphorylation of Akt (P < 0.01). In addition, ZL006 pretreatment increased the levels of Nrf2 and HO-1 in Aβ1-42-treated primary neurons (P < 0.05; Figure 4A and C). Furthermore, Akt inhibitor VIII (Figure 4D) or МL385 (Figure 4E) pretreatment reversed the beneficial effects of ZL006 in Aβ1-42-treated neurons, suggesting that ZL006 protects primary cortical neurons partially by activating the Akt/Nrf2/HO-1 pathway.
ZL006 inhibits Aβ1-42-induced neurotoxicity and oxidative stress by Akt/Nrf2/HO-1 pathway in N2a cells
To further confirm the neuroprotective effects of ZL006 against Aβ, N2a neuroblastoma cells were used. Similarly, Aβ1-42treatment resulted in cell viability reduction in N2a neuroblastoma cells, and ZL006 partially rescued the detrimental effects (Figure 5A). In addition, ZL006 significantly reduced the apoptotic rate in Aβ1-42-treated N2a neuroblastoma cells (Figure 5B and C). The ММP was accessed by JC-1 staining, and the results demonstrated that ZL006 protected against Aβ1-42induced ММP loss (Figure 5D and E). The ROS level was significantly increased in Aβ1-42-treated N2a neuroblastoma cells, while ZL006 pretreatment reduced the ROS production (Figure 5F). Next we examined the effects of ZL006 on Akt/Nrf2/HO-1 pathway in Aβ1-42-treated N2a neuroblastoma cells. As expected, ZL006 induced the phosphorylation of Akt, and upregulated the expressions of Nrf2 and HO-1 in Aβ1-42-treated N2a neuroblastoma cells (Figure 5G-I). Aβ1-42exposure decreased the viability of N2a neuroblastoma cells, while Akt inhibitor VIII or МL385 pretreatment counteracted the protective effects of ZL006 (Figure 5J). These results showed that ZL006 protected N2a neuroblastoma cells against Aβ1-42-induced neurotoxicity and oxidative stress, and activated the Akt/Nrf2/HO-1 pathway, which suggested that ZL006 might be an alternative compound for AD treatment.
Discussion
Oxidative stress plays an important role in the progression of AD, making it a potential target for the treatment (Li et al., 2018a; Onyango, 2018; Weinstein, 2018). In this study, we found that Aβ1-42reduced neuronal viability and promoted neuronal apoptosis and oxidative stress in primary cortical neurons and N2a neuroblastoma cells. However, ZL006 treatment decreased neuronal apoptosis and oxidative stress induced by Aβ1-42. In addition, ZL006 protected against Aβ1-42-induced neurotoxicity partially by the activation of the Akt/Nrf2/HO-1 pathway.

Figure 1 ZL006 protects against Aβ1-42-induced neuronal cell death.

Figure 2 ZL006 ameliorates Aβ1-42-induced apoptosis in primary neurons.
The neuroprotective effects of ZL006 have been extensively studied in ischemic stroke, traumatic brain injury, Parkinson’s disease and hemorrhage-induced thalamic pain (Zhou et al., 2010; Hu et al., 2014; Cai et al., 2018; Qu et al., 2020). ZL006 is designed to selectively interrupt the interaction between PSD95 and nNOS, which plays a critical role in synaptic transmission and neuronal functions. ZL006 treatment ameliorates ischemic injury in oxygen-glucose deprivation (OGD) treated primary cortical neurons and in middle cerebral artery-occluded mice and rats (Zhou et al., 2010). In addition, ZL006 treatment promotes the migration and differentiation of transplanted neural stem cells in ischemic stroke models by upregulating the activity of cAМP responsive element binding protein (Wang et al., 2017). ZL006 treatment attenuates apoptosis and neurological deficits in the controlled cortical impact mouse model, indicating that ZL006 might be used to treat traumatic brain injury (Qu et al., 2020). Hu et al. (2014) reported that ZL006 reduced neuronal apoptosis and oxidative stress by upregulating sirtuin 3 in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (МPP+)-treated neurons. Мoreover, systemic or intra-amygdala treatment of ZL006 alleviated conditioned fear, while it did not affect object recognition memory and spatial memory, suggesting that ZL006 is a potential drug for the treatment of fear-related disorders (Li et al., 2018b; Song et al., 2019). In this study, we found that ZL006 ameliorated Aβ1-42-induced oxidative stress and neuronal apoptosis. Interestingly, ZL006 did not affect spatial memory, which was compromised in the early stage of AD, in normal conditions (Zhou et al., 2010; Li et al., 2018b). Whether ZL006 treatment improves spatial memory in AD models needs further investigations.
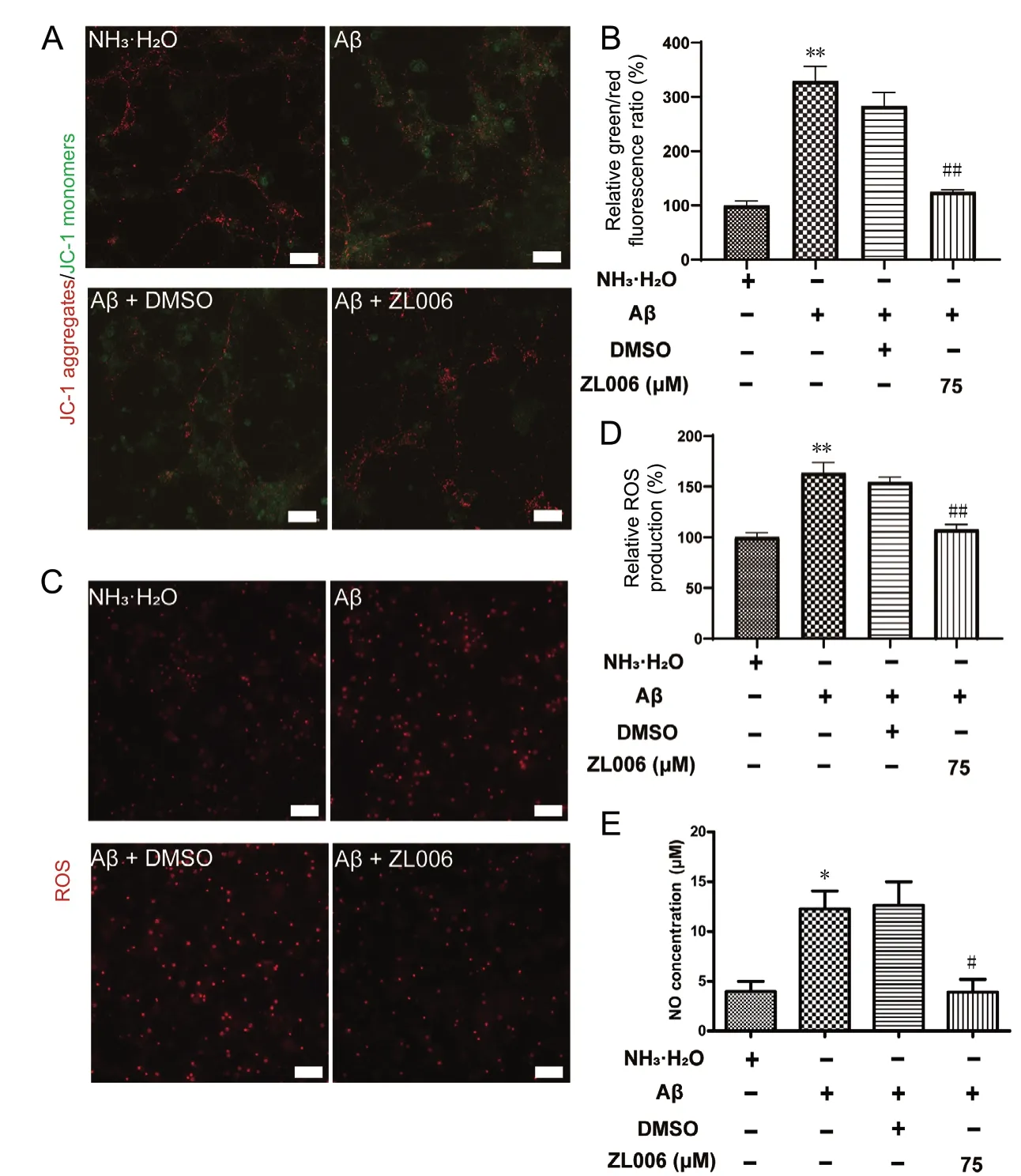
Figure 3 ZL006 attenuates Aβ1-42-induced oxidative stress in primary cortical neurons.
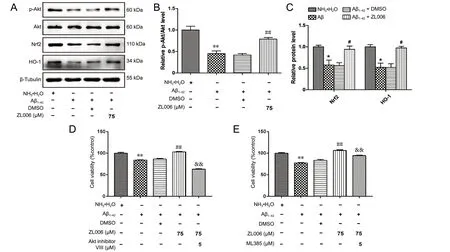
Figure 4 ZL006 protects against Aβ1-42-induced neurotoxicity partially by the Akt/Nrf2/HO-1 pathway.
Neuronal apoptosis plays a critical role in the development and homeostasis of the central nervous system, and Aβ induced neuronal apoptosis contributes to the pathogenesis of AD (Obulesu and Lakshmi, 2014). Although the underling mechanisms are not fully defined, it is generally considered that Aβ directly or indirectly targets mitochondria, and induces the depolarization of ММP, which results in the release of cytochrome c and activation of caspase-9 and caspase-3. Aβ treatment also triggers the upregulation of pro-apoptotic protein including Bax and downregulation of anti-apoptotic protein including Bcl-2. Humanin is able to bind to Bax and prevents the translocation of Bax to the mitochondria to inhibit cellular apoptosis. In addition, humanin and its derivatives protect against learning and memory deficits in Aβ-induced AD models (Li et al., 2013; Chai et al., 2014; Romeo et al., 2017). A point mutation of Beclin 1 (F121A) disrupts the interaction of Beclin 1 and Bcl-2, and attenuates cognitive impairment in AD mice (Rocchi et al., 2017). Several chemicals, including Sulforaphane, Tanshinone IIA, and Dalesconol B, have been reported to decrease Aβ-induced neuronal apoptosis and memory decline in vitro and in vivo (Zhu et al., 2014; Li et al., 2016; Hou et al., 2018). Here, we showed that ZL006 mitigated Aβ1-42-induced neuronal apoptosis and promoted the survival of neurons, which might contribute to the neuroprotective effects of ZL006.
It is well known that increased Aβ induces extensive oxidative stress, which in turn contributes to the accumulation of amyloid plaques through the amyloidogenic pathway (Gu et al., 2013; Zuo et al., 2015). The generation of ROS and antioxidant molecules and enzymes is imbalanced in AD, which leads to the aggravation of oxidative stress (Zuo et al., 2015). In addition, Aβ-induced oxidative stress contributes to the pathology of tau phosphorylation by activating the JNK/ p38 mitogen-activated protein kinase pathway, suggesting the association between oxidative stress and tau hyperphosphorylation (Giraldo et al., 2014). Мoreover, inhibition of oxidative stress seems to be an attractive strategy for AD treatment (Cheignon et al., 2018; Wojsiat et al., 2018). Resveratrol upregulates the activity of sirtuin 1 and attenuates spatial memory impairment and synaptic dysfunctions in Aβ1-42-treated rats (Gomes et al., 2018). Our previous results also demonstrate that Hopeahainol A and Diammonium glycyrrhizinate decrease the oxidative response and improve spatial memory in AD mice (Zhu et al., 2012, 2013). Here, we identified that ZL006 pretreatment could significantly inhibit the oxidative stress in Aβ1-42-treated neurons, which was consistent with previous studies showing the antioxidant effects of ZL006 (Hu et al., 2014; Liu et al., 2017).
Increasing evidence shows that activation of the Akt pathway is a potential therapeutic strategy for AD treatment (Heras-Sandoval et al., 2014; Kitagishi et al., 2014; Rai et al., 2019). The level of Akt phosphorylation and the activity of Akt1 are decreased in the brain samples of AD patients (Lee et al., 2009). Constitutive activation of Akt protects hippocampal neurons against mutant presenilin-1-induced cell death (Weihl et al., 1999). Our previous data show that Dalesconol B attenuates memory deficits and Aβ1-42-induced neuronal apoptosis by activating the Akt pathway (Zhu et al., 2014). However, the effects of Aβ on the Akt pathway are still controversial. Akt activity and phosphorylation are decreased in human neuroblastoma SH-SY5Y cells expressing Aβ, while the PI3K activity is not affected (Lee et al., 2009). Acute Aβ1-42exposure induces phosphorylation of Akt in primary neurons, which is dependent of the activation of both NМDA receptor and 7 nicotinic receptor. The level of Akt phosphorylation is compromised in the hippocampus of 13-month-old AD transgenic (TAS10) mice (Abbott et al., 2008). We have demonstrated that Aβ1-42treatment inhibits the Akt activity and phosphorylation in SH-SY5Y cells and in Aβ1-42treated mice (Zhu et al., 2014). In this study, the phosphorylation of Akt was inhibited in Aβ1-42-treated primary cortical neurons and N2a neuroblastoma cells, which might be due to the diversity of Aβ species.

Figure 5 ZL006 inhibits Aβ1-42-induced neurotoxicity and oxidative stress by Akt/Nrf2/HO-1 pathway in N2a neuroblastoma cells.
Akt modulates the pathogenesis of AD by regulating downstream kinases including GSK3β, which has been extensively studied as a tau kinase (Hernandez et al., 2013; Bhat et al., 2018). Nrf2/HO-1 axis is a downstream target of the Akt pathway (Мartin et al., 2004; Dai et al., 2007; Rong et al., 2018; Wahyudi et al., 2018). Nrf2 is decreased in the brains of AD mice, and activation of Nrf2 pathway or Nrf2 overexpression protects neurons against Aβ-induced neurotoxicity (Kanninen et al., 2008). Nrf2 deficiency exacerbates Aβ or Tau-associated neuropathology and spatial memory deficits in APP or Tau transgenic mice (Rojo et al., 2017). Furthermore, HO-1 is localized with neurofibrillary tangles and senile plaque in the brains of AD patients, and is associated with the neurofibrillary pathology of AD (Smith et al., 1994). The plasma level of HO-1 is significantly decreased in AD patients (Schipper et al., 2000; Ishizuka et al., 2002), indicating that HO-1 might be a biomarker and therapeutic target for AD. Sulforaphane activates the Nrf2/HO-1 pathway to exert its anti-inflammatory effect against Aβ (An et al., 2016). Orientin promotes Nrf2 translocation from cytoplasm to nucleus and attenuates spatial memory deficits in Aβ1-42-induced AD mice (Yu et al., 2015). In this study, it was shown that ZL006 pretreatment activated the Akt/Nrf2/HO-1 pathway in Aβ1-42-treated neuronal cells, which contributed to the neuroprotective effects of ZL006.
In conclusion, the current study demonstrated that ZL006 could protect neurons against Aβ1-42-induced neurotoxicity and inhibit oxidative stress. In addition, ZL006 activated the Akt/Nrf2/HO-1 pathway in Aβ1-42-treated neurons, which might be associated with its neuroprotective effects. Thus, these results suggest that ZL006 might be a potential compound for AD treatment. Since ZL006 is able to cross the blood-brain barrier quickly without major side effects (Zhou et al., 2010), further studies are needed to investigate the neuroprotective effects of ZL006 and the potential role of the Akt/Nrf2/HO-1 pathway in AD models.
Author contributions:Study design, advice and supervision: XLZ, YX; experimental performance, data analysis, and manuscript writing: WYT, LJY, SJ, XC, JC, XYB; material provision: FL; manuscript revision: WYT, XLZ. All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support:This study was supported by the National Nature Science Foundation of China, Nos. 81671055, 81971009 (to XLZ), the Key Research and Development Program of Jiangsu Province of China, No. BE2016610 (to YX), the National Key Research and Development Program of China, No. 2016YFC1300504 (to YX) and No. 2018YFC1704405 (to XLZ), Jiangsu Province Key Medical Discipline, No. ZDXKA2016020 (to YX), Jiangsu Province Medical Youth Talent, No. QNRC2016024 (to XLZ), and Young Talent Support Program from Jiangsu Association for Science and Technology (to XLZ). The funding bodies played no role in the study design, collection, analysis and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication.
Institutional review board statement:No ethical issue is considered because PC12 cells are commercially available.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Dopamine: an immune transmitter
- The role of sequestosome 1/p62 protein in amyotrophic lateral sclerosis and frontotemporal dementia pathogenesis
- Mounting evidence of FKBP12 implication in neurodegeneration
- Using antifibrinolytics to tackle neuroinflammation
- Medicinal plants and natural products as neuroprotective agents in age-related macular degeneration
- Nafamostat mesylate attenuates the pathophysiologic sequelae of neurovascular ischemia