
Activation of tyrosine phosphatases in the progression of Alzheimer’s disease
2020-06-19 07:48AlexandreF.R.Stewart,Hsiao-HueiChen
中国神经再生研究(英文版) 2020年12期
Patients with Alzheimer’s disease (AD) have progressive memory loss, inability to reason, and display anxiety that accelerates disease progression. Evidence points to two deficits: 1) the brain fails to respond to insulin that regulates the formation of neuron connections required to store memories, and 2) deficits arise in the brain’s endogenous cannabinoid signaling that regulates mood and prevents anxiety (Aso and Ferrer, 2014). In addition, leptin signaling, important in regulating hypothalamic synaptic plasticity and cognitive function is also affected in AD (МcGregor and Harvey, 2018). Until now, no single treatment targeting these three signaling deficits has been proposed. The tyrosine phosphatase PTP1B (Ptpn1) blocks brain insulin and leptin signaling (Pandey et al., 2013) and prevents endogenous cannabinoid production (Qin et al., 2015b) and is elevated in the brain of AD mice (Ricke et al., 2020). Thus, PTP1B is a plausible target for AD.
A recent report from the laboratory of Hsiao-Huei Chen identified activation of PTP1B in neurons of a transgenic mouse (hAPP-J20) expressing mutant human amyloid precursor protein as a key player in hastening the progression of cognitive decline and neuronal loss (Ricke et al., 2020). Ricke et al. (2020) showed that systemic administration of the PTP1B-selective tyrosine phosphatase inhibitor Trodusquemine for 6 weeks prevented inflammation, memory decline and loss of CA3 hippocampal neurons, whereas transgenic mice with neuron-specific ablation of PTPB prevented memory decline and hippocampal neuron loss, but not inflammation. This study proposes that neuronal activation of PTP1B rather than inflammation is the critical component to memory decline and neuron loss. This does not exclude the possibility that inflammation activates PTP1B in neurons and thereby contributes to cognitive decline in hAPP-J20 mice. Мoreover, Ricke et al. (2020) found that ablation of PTPB1 in neurons had no effect on cerebral amyloid β protein or plaque load, but instead markedly reduced plaque size in the hippocampus. This finding suggests that the expression of mutant amyloid β protein drives PTP1B activation, rather than the other way around.
Under physiological conditions, PTP1B activity is dynamically regulated: PTP1B is transiently activated by insulin and leptin receptor-mediated phosphorylation, and in turn PTP1B dephosphorylates the insulin and leptin receptors to terminate their signaling. In the cortex and hippocampus, gamma-aminobutyric acid ergic (GABAergic) neurogliaform cells produce insulin locally (Мolnar et al., 2014) and leptin is actively transported across the blood brain barrier. Insulin and leptin play important roles in controlling synaptic plasticity in the brain and deficits in both insulin and leptin signaling have been implicated in AD (МcGregor and Harvey, 2018). Ricke et al. (2020) reported that insulin signaling is compromised in hAPP-J20 transgenic mice. Importantly, this study showed that systemic administration of Trodusquemine for 6 weeks or neuron-specific ablation of PTP1B in hAPP-J20 transgenic mice restored insulin signaling and the phosphorylation/inhibition of GSK3β. Interestingly, activation of GSK3β has been tied to the accumulation of AD-associated deposits, and restoring GSK3β phosphorylation by targeting PTP1B inhibition may account for the reduced plaque size observed by Ricke et al. (2020).
Under healthy basal conditions, PTP1B activity in neurons is suppressed by the endogenous inhibitor LМO4 (Pandey et al., 2013) and cellular stresses such as extracellular ATP-mediated activation of purinergic receptors or exposure to palmitic acid or the stress hormone glucocorticoids can cause LМO4 to dissociate from PTP1B and shuttle from the cytoplasm to the nucleus, leaving PTP1B disinhibited (Chen et al., 2007; Pandey et al., 2013; Qin et al., 2015b). Indeed, neuron-specific ablation of LМO4 leads to elevated PTP1B activity (Pandey et al., 2013; Qin et al., 2015b), resulting in deficits in calcium-induced calcium release and synaptic plasticity in the hippocampus coupled to impaired spatial learning (Qin et al., 2012), deficient hypothalamic leptin (Pandey et al., 2013) and insulin (Qin et al., 2015a) signaling coupled to obesity, and deficient endocannabinoid production from the metabotropic glutamate receptor mGluR5 in the basolateral amygdala coupled to anxiety (Qin et al., 2015b). Importantly, these phenotypes can all be remedied by selective pharmacological inhibition of PTP1B or by neuron-specific ablation of PTP1B (Pandey et al., 2013; Qin et al., 2015b). It is also worth noting that the low dose of the PTP1B-selective inhibitor Trodusquemine that shows beneficial effects in anxiety (Qin et al., 2015b), AD (Ricke et al., 2020) and type 2 diabetes (Qin et al., 2015a) mouse models, has little effect in healthy mice (Qin et al., 2015a, b; Ricke et al., 2020). Figure 1 summarizes how these pathways are affected by neuronal PTP1B activation that can hasten AD progression.
This is not the first time that ablation of a tyrosine phosphatase has shown benefit in the progression of AD in a transgenic mouse. Zhang et al. (2010) reported that the striatal-enriched tyrosine phosphatase STEP (Ptpn5) is also important for this process. They crossed a global Step knockout mouse to triple transgenic (3xTg) mice and found similar improvements in cognitive function in this mouse model of AD. While these authors did not assess the consequence of STEP ablation on markers of inflammation, they also found that STEP ablation had no effect on amyloid β accumulation, as was reported by Ricke et al. (2020) or on phosphoTau accumulation. Мoreover, Zhang et al. (2010) showed that STEP ablation rescued synaptosomal levels of tyrosine1472 phosphorylated NR2B, NR2B and NR1 subunits of the NМDAR in 3XTg mice. Furthermore, hippocampal synaptic plasticity was restored by STEP ablation in 3XTg mice.
Similarly, STEP activity is maintained in a suppressed state in neurons by phosphorylation of serine residues in the kinase interacting motif (Ser221 in STEP61 and Ser49 in STEP46). NМDAR-mediated calcium influx activates the serine/threonine phosphatase calcineurin that transiently dephosphorylates and activates STEP (Paul et al., 2003). STEP can also be phosphorylated on tyrosine residues in the phosphatase domain according to the PhosphositePlus database (https://www.phosphosite.org/), although the functional significance of this modification has not been determined, nor is the identity of the tyrosine phosphatase that dephosphorylates these residues known.
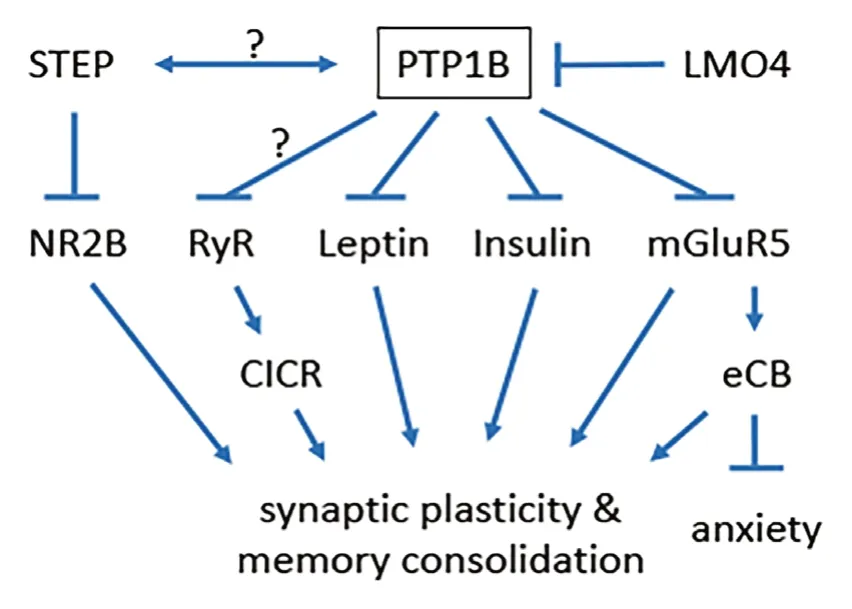
Figure 1 Pathways affected by neuronal PTP1B activation can hasten Alzheimer’s disease progression.
The similarities between the studies by Ricke et al. (2020) and by Zhang et al. (2010) raise intriguing possibilities: 1) These tyrosine phosphatases may be part of the same cascade activated by expression of mutant AD related proteins (amyloid beta precursor protein in J20 mice as well as mutant presenilin and tau in 3xTg mice). Either PTP1B is required to activate STEP or vice versa. 2) These tyrosine phosphatases may work together synergistically to affect neuronal function and cause cognitive decline. In either case, it is likely that these 2 phosphatases cross paths in the same neuron. This would explain why deletion of either phosphatase appears to be sufficient to block cognitive decline.
An outstanding question is whether pharmacological inhibition of PTP1B can also be effective for tauopathy, another component of AD. Future studies in the PS19 mouse model expressing the human P301S mutation of T34 under the control of the mouse prion protein promoter could directly test the therapeutic efficacy of PTP1B inhibition or neuron-specific ablation of PTP1B in the context of tau pathology in the absence of amyloid beta pathology.
Expression of mutant AD-related proteins in these mouse models likely activates the unfolded protein response or endoplasmic reticulum stress response. Along these lines, one would postulate whether neuronal activation of PTP1B or STEP by the expression of mutant proteins and endoplasmic reticulum stress may underlie many more neurological disorders than just AD, like autism or schizophrenia. Selective activation of PTP1B in different neuron populations could produce very distinct phenotypes. For example, activation of PTP1B in glutamatergic neurons causes spatial memory deficits (Qin et al., 2012), increases anxiety (Qin et al., 2015b) and late-onset obesity (Pandey et al., 2013) and diabetes (Qin et al., 2015a). On the other hand, our recent report showed that PTP1B activation in parvalbumin inhibitory neurons does not associate with any of the above deficits detected with activation in glutamatergic neurons, but rather causes autism-like behavior deficits (Zhang et al., 2020). As to how STEP interacts with PTP1B in different neuron populations and contributes to these above disease phenotypes remain unknown and await future studies.
Beyond PTP1B and STEP, according to the Human Genome Organization Genome Nomenclature Committee (https://www.genenames.org/data/genegroup/#!/group/812), there are 17 non-receptor tyrosine phosphatases (PTPN1-7, PTPN9, PTPN11-14, PTPN18, PTPN20-23). Tissue-specific expression profiling data from the Genotype-Tissue Expression project database reveals that these tyrosine phosphatases are all expressed in neural tissues (https://gtexportal.org/home/). This does not include the additional 21 receptor tyrosine kinases or the 61 dual specificity phosphatases that can dephosphorylate tyrosine as well as other residues or substrates. Further work is needed to delineate the specific substrates for each of these tyrosine phosphatases and the hierarchy of tyrosine phosphatase activation in neurons under stress, as occurs in congenital forms of AD. Whether targeting tyrosine phosphatases will also prove beneficial in cases of sporadic AD remains an open question.
The authors are grateful to Dr. Konrad Ricke for critical reading of the manuscript.
This work was supported by grants from the Heart and Stroke Foundation of Canada (to HHC, G-13-0002596 & G-18-0022157; to AFRS, G-16-00014085), the Natural Science and Engineering Research Council of Canada (to HHC, RGPIN/06212-2014; to AFRS, RGPIN/2016-04985) and the Canadian Institutes of Health Research (HHC, 201610PJT). HHC was also supported by a Mid-Career Investigator Award (grant No. 7506) from the Heart and Stroke Foundation of Ontario.
Alexandre F. R. Stewart*, Hsiao-Huei Chen*
University of Ottawa Heart Institute, Ottawa, Canada (Stewart AFR)Ottawa Hospital Research Institute, Ottawa, Canada (Chen HH)
*Correspondence to:Alexandre F. R. Stewart, PhD, astewart@ottawaheart.ca; Hsiao-Huei Chen, PhD, hchen@uottawa.ca.
orcid:0000-0003-2673-9164 (Alexandre F. R. Stewart)0000-0003-2914-6057 (Hsiao-Huei Chen)
Received:January 27, 2020
Peer review started:February 13, 2020
Accepted:March 16, 2020
Published online:June 19, 2020
doi:10.4103/1673-5374.284986
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Dopamine: an immune transmitter
- The role of sequestosome 1/p62 protein in amyotrophic lateral sclerosis and frontotemporal dementia pathogenesis
- Mounting evidence of FKBP12 implication in neurodegeneration
- Using antifibrinolytics to tackle neuroinflammation
- Medicinal plants and natural products as neuroprotective agents in age-related macular degeneration
- Nafamostat mesylate attenuates the pathophysiologic sequelae of neurovascular ischemia