
核磁共振定量法测定盐酸莫西沙星的含量
2020-06-17 13:30秦玲萍王新仙卢定强
生物加工过程 2020年3期
秦玲萍,王新仙,卢定强,
(1.南京工业大学 药学院,江苏 南京 211800;2.江苏省药物研究所有限公司,江苏 南京 210009)
喹诺酮类是一类较新的合成抗菌药,主要通过阻碍细菌的DNA螺旋酶(维系染色体的负超螺旋形式),使mRNA和蛋白质的合成不受DNA控制,进而阻断DNA的复制,使其产生杀菌作用,对多种革兰氏阴性菌也有杀菌作用,被广泛应用于泌尿生殖系统疾病、胃肠疾病,以及呼吸道、皮肤组织受到革兰阴性细菌感染的相关治疗中[1-3],并与其他抗菌药之间不存在交叉耐药性。
盐酸莫西沙星(moxifloxacin hydrochloride,CAS 186826-86-8),化学名为1-环丙基-7-{S,S-2,8-重氮-二环[4.3.0]壬-8-基}-6-氟-8-甲氧-1,4-二氢-4-氧-3-喹啉羧酸氢氯化物,是1999年由德国拜耳公司研制的第四代超广谱喹诺酮类药物。作为第四代喹诺酮类的典型代表药物,盐酸莫西沙星的特点就是在结构中引入8-甲氧基,从而有利于提高抗厌氧菌活性,在C7位上的氮双杂环结构则能增强抗革兰阳性菌的活性,并且可以维持原有的抗革兰阴性菌的活性效果,因此很少有恶心、头痛及精神错乱等副作用发生。在临床上通过对照实验发现:盐酸莫西沙星与克拉霉素相比,在治疗慢性支气管炎方面效果更佳,能有效提高细菌清除率[4-5]。在治疗社区获得性肺炎及老年下呼吸道感染等疾病的临床治疗效果、细菌清除率均高于头孢呋辛,不良反应少[6-7]。对于抗感染能力也强于左氧氟沙星[8-10]。由此看来,盐酸莫西沙星未来将逐渐占领抗生素市场。
目前文献报道有采用HPLC法,也有采用紫外或非水滴定的方法测定盐酸莫西沙星含量的研究[11-13],但滴定法重现性较差、精密度不高,给操作过程增加了难度;HPLC法则须要配缓冲液等操作,费时费力,会给结果带来不确定性以及灵敏度不高等问题;测定含量时,对照品较难获得也是一个重要因素;而且必须测定分析物水分含量、炽灼残渣和残留溶剂等才能得出绝对含量;同时,这些方法测定含量的前提是要有盐酸莫西沙星的对照品。因此,建立一种快速方便、精密度高以及专属性强且准确的含量测定方法对盐酸莫西沙星的研究有着重大意义。
目前,核磁共振技术越来越多地应用于定量分析领域[14-17],已先后被美国药典、欧洲药典和中国药典2015版收录。核磁定量分析的方法有2种,分别是内标法和外标法,现在较为常用的是内标法,也叫做绝对测量法,该方法可以直接快速地测量物质的含量。与其他分析方法比较,核磁定量内标法的最大优势就在于只需用已知纯度的化合物作参比,即使在没有对照品的情况下也能快速准确并且不破坏样品结构以测定含量,同时该方法也可作为日常分的工作中标化工作对照品的方法,而用于日常分析的测定也较为简单方便快捷。
本研究中,笔者采用氢核磁共振定量法(qHNMR)和氟核磁共振定量法(qFNMR)来测定盐酸莫西沙星的含量,并将其结果与传统的质量平衡法测定结果进行比较,以期实现两种结果相对一致,从而为盐酸莫西沙星的含量测定提供一种简单、快速、准确且可常规使用的检测方法。
1 材料与方法
1.1 仪器
400 MHz AVANCE型核磁共振谱仪,瑞士Bruker公司;LC-2010A型高效液相色谱仪,日本岛津有限公司;MS-100型水分自动测定仪,上海佳实电子科技有限公司。
1.2 试剂
盐酸莫西沙星原料药(批号:20171204、20180109和20181211),江苏永达药业有限公司;D2O(质量分数≥99.8%),上海梯希爱化成工业发展有限公司;3,5-二甲基吡唑(质量分数≥99%),上海阿拉丁生化科技股份有限公司;三氟乙酸(质量分数≥99.5%),上海麦克林生化科技有限公司;其余试剂均为国产分析纯。
1.3 定量氢核磁共振法
1.3.1 定量氢核磁共振法的测试条件
采用 zg30 脉冲序列,测定温度25 ℃,脉冲宽度(P1)14.8 s,样品扫描次数(NS)16次,采样时间(AQ)为4.0 s,弛豫时间(D1)为15 s。
1.3.2 定量氢核磁共振法的供试品溶液的配制
精密称取盐酸莫西沙星原料药17.89 mg及3,5-二甲基吡唑11.95 mg,加入1 mL D2O,超声溶解、混匀、过滤,转移0.5 mL至5 mm核磁管中,进核磁仪检测。
1.3.3 定量氢核磁共振法的对照品溶液的配制
精密称取3,5-二甲基吡唑11.86 mg于10 mL容量瓶中,得到质量浓度为1.186 mg/mL的3,5-二甲基吡唑内标溶液。用该内标溶液进行稀释,分别配制质量浓度为1.01、1.47、2.03、2.84和3.88 mg/mL的盐酸莫西沙星溶液。之后进行超声溶解,混匀,放置待测。
1.4 定量氟核磁共振法
1.4.1 定量氟核磁共振法的测试条件
采用 zgfhigqn. 2脉冲序列,测定温度25 ℃,谱宽(SWH)12 500 Hz,采样次数(NS)16次,采集时间(AQ)5.24 s,弛豫时间(D1)10 s。
1.4.2 定量氟核磁共振法的供试品溶液的配制
精密称取盐酸莫西沙星原料药15.72 mg及三氟乙酸17.48 mg,加入1 mL D2O超声溶解,混匀、过滤、转移0.5 mL至5 mm核磁管中,进核磁仪检测。
1.4.3 定量氟核磁共振法的对照品溶液的配制
精密称取三氟乙酸22.46 mg于10 mL容量瓶中,得到内标溶液质量浓度为2.246 mg/mL。用该内标溶液进行稀释,配制盐酸莫西沙星溶液,质量浓度分别为1.05、1.51、2.12、2.92和4.01 mg/mL。进行超声溶解、混匀、放置待测。
2 结果与讨论
2.1 核磁定性的分析结果
盐酸莫西沙星的1H NMR分析结果见表1。由表1可知:盐酸莫西沙星中3个H原子为活泼氢,均在D2O中消失。氟谱仅在化学位移δ=120.06处出现一个单峰(δ=75.66处为内标峰)。1H NMR和19F NMR图谱结果与盐酸莫西沙星分子式(C21H24FN3O4·HCl)相符,其结构式见图1,盐酸莫西沙星的氢谱(图2)共显示22个质子峰。
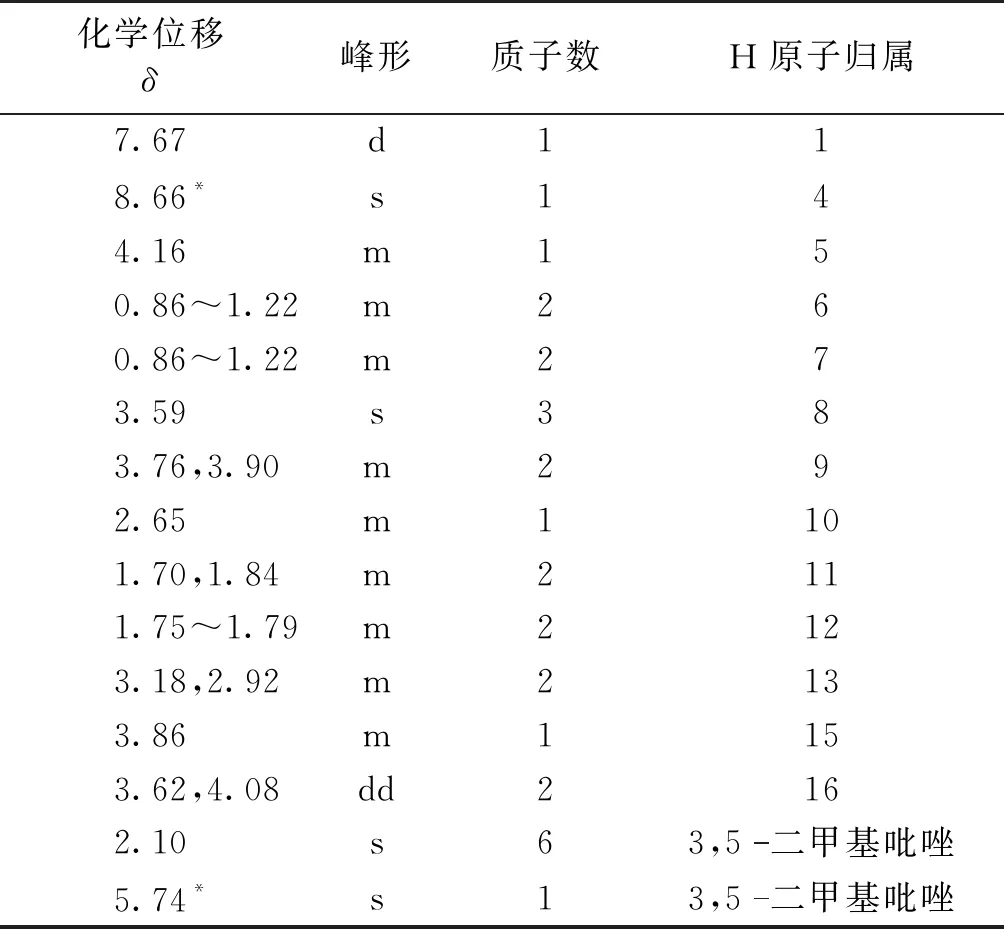
表1 盐酸莫西沙星的1H NMR解析
注:*为定量峰;d为双峰;s为单峰;m为多重峰;dd为双二重峰。
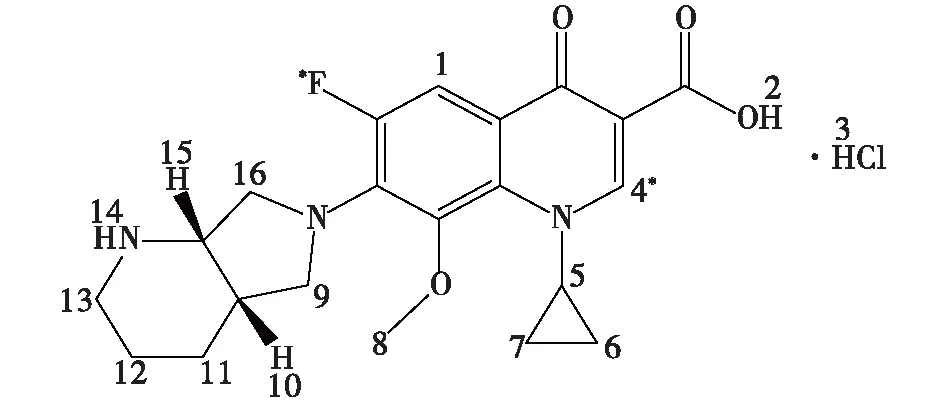
图1 盐酸莫西沙星结构Fig.1 Structure of moxifloxacin hydrochloride
2.2 定量氢核磁共振法测定盐酸莫西沙星的含量
2.2.1 方法学研究
1)专属性研究
按1.2项中盐酸莫西沙星溶液进行测样,得到盐酸莫西沙星与3,5-二甲基吡唑混合溶液的1H NMR(图2)。从图2可以看出:盐酸莫西沙星δ=8.66的信号峰与3,5-二甲基吡唑δ=5.74的信号峰能够完全分离,互不影响,专属性较强。
2)精密度试验
分别平行制备6份供试品溶液,进样测定,计算得到盐酸莫西沙星的含量,测得结果的相对标准偏差(RSD)为1.2%。
3)准确度试验
精密配制样品及内标溶液,平行制备9份,平均分成3组,各组精密加入盐酸莫西沙星标准溶液0.1、0.2、0.3 mL,测定并以内标法计算加标样品的含量。通过实际测得加入量与理论添加量的比值计算回收率,结果表明,平均回收率范围是98.86%~99.70%,相对标准偏差(RSD)为0.3%。
4)稳定性试验
将盐酸莫西沙星供试品溶液,分别放置于0、2、4、8和12 h后进样测定。以其定量峰与内标峰面积的比值为衡量,计算得RSD为0.59%,数据结果表明:样品溶液在测试环境中12 h内稳定。
5)线性范围试验
取1.3.3项中用内标溶液稀释配制的质量浓度为1.01、1.47、2.03、2.84和3.88 mg/mL的盐酸莫西沙星溶液0.5 mL至核磁管中,进核磁仪检测,得到盐酸莫西沙星与内标的1H NMR谱。以配制的质量浓度(X)为横坐标,盐酸莫西沙星定量峰与内标峰峰面积之比(Y)为纵坐标进行线性回归。得到回归方程为Y=1.387X+0.512 8(R2= 0.999 4),说明在1.01~3.88 mg/mL的质量浓度范围内,盐酸莫西沙星溶液质量浓度与其定量峰内标峰面积比呈线性关系。
6)定量限与检测限
取盐酸莫西沙星溶液,进行稀释检测,以信噪比(S/N)为10时对应的质量浓度为定量限,以信噪比(S/N)为3时对应的质量浓度为检测限。结果显示:使用定量氢核磁共振法测定盐酸莫西沙星含量时,定量限为0.268 mg/mL,检测限为0.089 mg/mL。

图2 盐酸莫西沙星与3,5-二甲基吡唑混合物的1H NMR谱图Fig.2 1H NMR spectrum of the mixture of moxifloxacin hydrochloride and 3,5-dimethylpyrazole
2.2.2 含量测定
分别精密称取3批盐酸莫西沙星原料药,制备进样溶液,按1.3.1项中的测试条件进样测定,并且通过式(1)计算盐酸莫西沙星的含量。
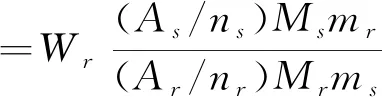
(1)
式中:As、Ar分别为盐酸莫西沙星定量峰和内标峰的峰面积;ns、nr分别为盐酸莫西沙星定量峰和内标峰所代表的原子数;Ms、Mr分别为盐酸莫西沙星和内标的相对分子质量;ms、mr分别为盐酸莫西沙星和内标的相对分子质量;Wr为内标的百分含量。
最后,分别得到3批盐酸莫西沙星含量为98.19%、98.07%和98.11%。
2.3 定量氟核磁共振法测定盐酸莫西沙星的含量结果
2.3.1 方法学研究结果
1)专属性研究结果
按1.4.2项下供试品溶液进行测样,得到盐酸莫西沙星与三氟乙酸的19F NMR图谱,结果见图3。由图3可知,19F NMR图谱仅有2个信号峰,分别是δ=120.08和δ=75.66,是盐酸莫西沙星和内标物三氟乙酸的信号峰,说明该方法能完全分离盐酸莫西沙星,专属性强。
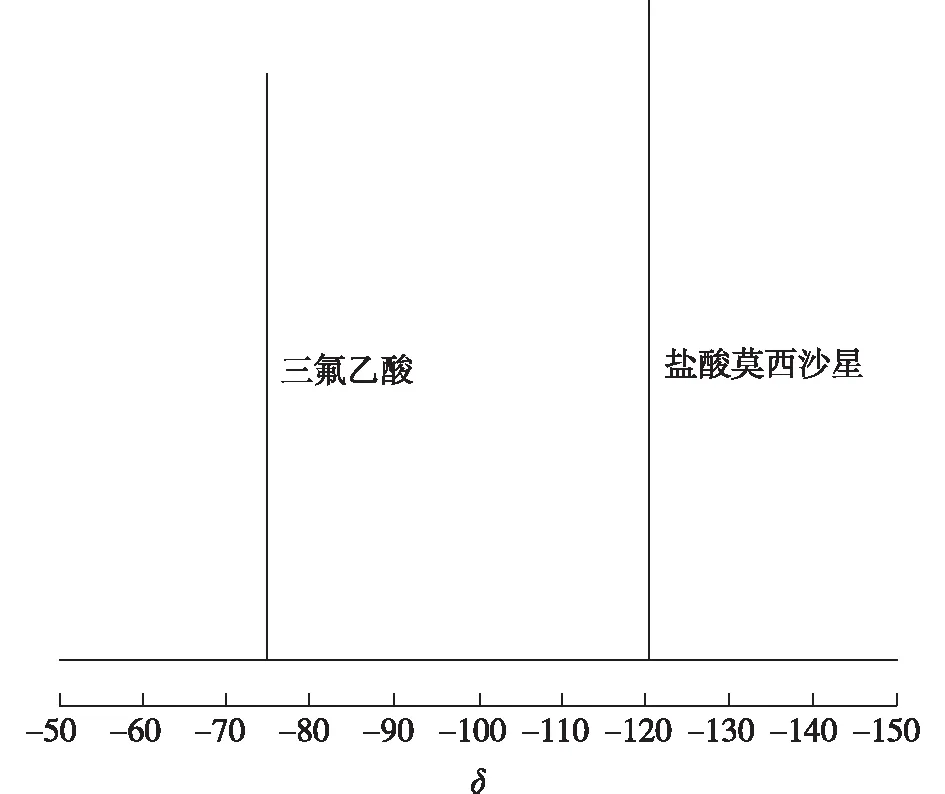
图3 盐酸莫西沙星与三氟乙酸混合物的19F NMR谱图Fig.3 19F NMR spectrum of the mixture of moxifloxacin hydrochloride and trifluoroacetic acid
2)精密度试验结果
分别平行制备6份供试品溶液,进样测定,计算盐酸莫西沙星的含量,测得结果的相对标准偏差(RSD)为1.1%。
3)准确度试验结果
精密配制样品及内标溶液,平行制备9份,平均分成3组,各组分别精密加入盐酸莫西沙星标准溶液0.1、0.2、0.3 mL,测定并以内标法计算加标样品的含量。通过实际测得加入量与理论添加量的比值计算回收率,结果表明:平均回收率范围是98.64%~100.41%,相对标准偏差(RSD)为0.5%。
4)稳定性试验结果
将盐酸莫西沙星供试品溶液,分别放置于0、2、4、8和12 h后进样测定。以其定量峰与内标峰面积的比值为衡量,计算得RSD为0.61%,数据结果表明样品溶液在测试环境中12 h内稳定。
5)线性范围试验结果
取1.4.3项中用内标溶液稀释配制的盐酸莫西沙星溶液,质量浓度为1.05、1.51、2.12、2.92和4.01 mg/mL,取0.5 mL至核磁管中,进核磁仪检测,得到盐酸莫西沙星的19F NMR谱。以配制的盐酸莫西沙星溶液质量浓度(X)为横坐标,盐酸莫西沙星定量峰与内标峰峰面积之比(Y)为纵坐标进行线性回归。得回归方程为Y=1.344 4X+0.504 2(R2=0.999 5),说明在1.05~4.01 mg/mL的质量浓度范围内,与其定量峰内标峰面积比呈线性关系。
6)定量限与检测限结果
取盐酸莫西沙星溶液,进行稀释检测,以信噪比(S/N)为10时对应的质量浓度为定量限,以信噪比(S/N)为3时对应的质量浓度为检测限。结果显示:使用定量氟核磁共振法测定盐酸莫西沙星含量时,定量限为0.087 mg/mL,检测限为0.029 mg/mL。
2.3.2 含量测定结果
分别精密称取3批盐酸莫西沙星原料药,制备进样溶液,按1.4.1项中测试条件进样测定,按2.2.2项中式(1)计算盐酸莫西沙星含量,分别得到3批盐酸莫西沙星含量为98.21%、98.09%和98.15%。
2.4 质量平衡法定值的结果
取3份不同批号的盐酸莫西沙星原料药,用HPLC进行测定,面积归一化法计算,得到结果分别为99.47%、99.84%和99.75%;水分测定结果分别为1.02%、1.01%和1.06%;炽灼残渣分别为0.11%、0.16%和0.12%;3批盐酸莫西沙星残留溶剂均为0.000 2%,根据质量平衡法得式(2)。
w(盐酸莫西沙星)=[100%-w(水分)-
w(炽灼残渣)-w(残留溶剂)]×HPLC测定含量
(2)
计算得3批的含量分别为98.33%、98.67%和98.57%。数据表明:1H NMR和19F NMR的测定结果与传统质量平衡法基本一致。
2.5 内标及溶剂的选择
内标物的选择在核磁定量实验中具有重要意义,并且选择受到限制,通常须要符合几个特定要求:①纯度较高;②在所选溶剂中有良好稳定性和溶解性;③NMR谱图中有清晰且分离的信号峰;④与分析物没有化学反应。
从图2可以看出:内标物3,5-二甲基吡唑的内标峰(δ=5.74)与盐酸莫西沙星定量峰(δ=8.66)互不干扰,可以较好地定量,所以选择3,5-二甲基吡唑作为定量氢谱的内标物。同理,定量氟谱中选择三氟乙酸也是较为合适的。在氘代试剂D2O中,分析物及内标物都有很好的溶解性和稳定性,溶剂峰也不影响定量,所以选择D2O为氘代试剂。
2.6 内标物与被测物定量峰的选择
笔者采用核磁技术对物质进行定量,在氢核磁定量的时候,往往会出现多个峰,此时选择定量峰的关键基于:①与周边的峰不重叠、互不干扰,附近基线平整;②最好选择单峰,在计算积分面积时更为准确。
因此,本文中的氢核磁定量选择了δ=5.74处单峰为内标峰,δ=8.66处单峰为分析物的定量峰,在定量计算时更为精准。
3 结论
本研究建立了2种核磁定量(1H NMR和19F NMR)的新方法来测定盐酸莫西沙星的含量,同时对该方法进行了方法学验证及考察,最后将测定计算结果与质量平衡法结果进行比较。结果表明:两种核磁定量技术的测定结果与质量平衡法基本一致,满足了对盐酸莫西沙星日常质量控制分析的要求,同时也为对照品的标定提供了一种新的测定方法。定量核磁具有操作简单,不需要分析物对照品,不会破坏分析物结构等优势。在高磁场的应用及傅立叶变换技术的不断发展下,核磁共振技术在定量分析领域中的应用会越来越广。
猜你喜欢
现代仪器与医疗(2022年4期)2022-10-08
农业工程学报(2022年6期)2022-06-27
科学导报(2022年4期)2022-01-26
今日农业(2021年2期)2021-11-27
今日农业(2020年23期)2020-12-31
含能材料(2020年7期)2020-07-11
学与玩(2017年11期)2017-02-16
山西青年(2017年2期)2017-01-11
百姓生活(2016年6期)2016-06-22
应用海洋学学报(2015年4期)2015-11-24
