
酮内酯药物结构修饰的研究进展
2020-06-12 01:33李亚鑫刘斯斯王风萍常桂凤
河北北方学院学报(自然科学版) 2020年8期
李亚鑫,刘斯斯,王风萍,常桂凤,李 炜
(河北北方学院药学系,河北 张家口 075000)
红霉素在临床上常用于治疗上下呼吸道感染,对青霉素过敏者及支原体肺炎患者红霉素治疗效果显著。红霉素结构中含有2个糖基基团:C-3位上的克拉定糖与C-5位上的德胺糖,其中德胺糖的羟基可与核糖体50S亚基上的A2058碱基在新生肽通道(NPET)形成氢键,使得红霉素可以牢固的与肽基转移酶中心(PTC)附近位点结合,发挥抑菌作用[1-2]。克拉霉素、阿奇霉素等第二代大环内酯类药物克服了红霉素酸不稳定性这一缺陷,提高了其在体内的生物利用度,提高了药代动力学性质。然而自20世纪90年代以来,大环内酯类抗生素滥用导致了细菌耐药性不断增强,尤其是耐甲氧西林金黄色葡萄球菌对亚洲国家的公共健康构成了极大威胁[3-4]。
早期研究认为,红霉素C-3位连接的克拉定糖是大环内酯类抗生素的药效团。但是AGOURIDAS和DENIS等人合成了一系列3-羰基红霉素衍生物,构效关系表明,红霉素C-3位的克拉定糖可以诱导细菌产生耐药性[5]。这一发现为红霉素结构修饰开辟了新的研究方向,此后第三代大环内酯类药物的研究方向重点围绕酮内酯展开。在大环内酯C-2位将氢原子替换为其生物电子等排体氟原子,可避免C-3位羰基烯醇式互变,有利于保持其四面体结构,提高其稳定性,扩展药物抗菌谱,优化药代动力学性质。我们查阅了近年来关于酮内酯尤其是2-氟酮内酯类药物相关研究的文献,比较文献中涉及的不同酮内酯结构与活性的关系,以期为酮内酯药物构效关系研究提供参考。
1 C-11,12位引入芳香侧链
DENIS等人进行构效关系研究后找到了一个C-11,12位连接吡啶咪唑杂环,C-3位修饰为羰基具有高活性新型大环内酯类抗生素HMR-3647,即目前酮内酯系列第一个上市药物——泰利霉素(图1)。泰利霉素与细菌核糖体复合物的高分辨率X射线晶体结构表明,泰利霉素C-11,12位上的芳香侧链可与二级结合位点A752发生π-π结合[6-7]。
泰利霉素与少见特异体质相关但易致死的肝毒性使得FDA限制了其适用范围,撤销了2个关于鼻窦炎、支气管炎适应症和皮肤感染适应症,仅剩下社区获得性细菌性肺炎适应症,这可能与泰利霉素引入的芳香侧链有关[8-9]。近期研究表明泰利霉素链接的芳香侧链中吡啶环可能是引起这种不良反应的原因[10]。不确定的潜在风险迫使药品生产商终止了泰利霉素相关衍生物的研发[11]。
Cempra公司研发了一种结构不同于泰利霉素的酮内酯—索利霉素(图1)。其中,C-2位引入了氟原子,C-3位修饰为羰基,C-11,12位引入了3-氨基苯基-[1,2,3]-三唑。体外活性测试结果表明,索利霉素对mef诱导的红霉素耐药肺炎链球菌抑菌活性是泰利霉素的8倍,对金黄色葡萄球菌的抑菌活性是泰利霉素的2倍,对红霉素耐药的流感嗜血杆菌的抑菌活性是泰利霉素的4倍[12-14]。目前索利霉素已进入临床研究阶段。
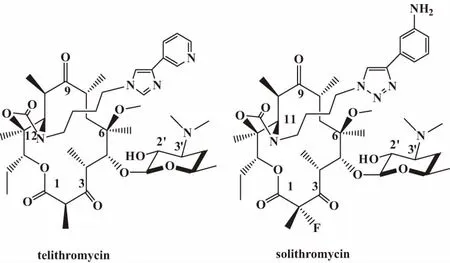
图1 telithromycin and solithromycin
KANEKO等人在C-12位选择性引入了叠氮基团,随后C-11,12位成环引入了不同芳香环,同时使用六甲基二硅基胺基钾(KHMDS)拔氢,使用亲电氟化试剂实现了C-2位氟化,得到的2-氟酮内酯系列化合物对化脓性链球菌S.pyogenes表现出良好的抗菌活性[15]。
2 C-6位引入芳香侧链
大环内酯C-6位羟基通过烯丙基或炔丙基修饰,可以通过Heck或Sonogashira反应偶联不同芳香侧链,避免了C-6位羟基与C-9位羰基反应的同时,可对耐药菌产生新结合靶点。
BEEBE等人以9-肟酮内酯为原料,用NaH拔氢,以N-氟代苯磺酰亚胺(NFSI)为氟代试剂,对C-2位进行氟化修饰,通过Sonogashira反应在C-6位偶联了2个不同芳杂环,合成2个2-氟-9-肟酮内酯化合物8b和8c(图2),同时合成了相应的C-2位未氟化9-肟酮内酯化合物5b和5c(图2)。2个系列化合物的体外活性测试结果表明,对化脓性链球菌S.pyogenes930,C-2位氟化系列化合物比未氟化系列化合物的最小抑菌浓度低4倍[16]。
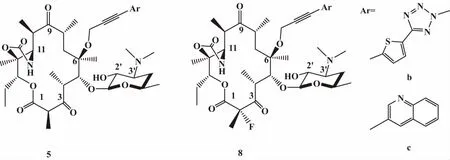
图2 Structure of compounds 5 and 8
PLATA等人开辟了一条新的合成路径,在红霉素的6位羟基通过烯丙基化、Heck反应引入3-喹啉基,2'位通过乙酰基保护后,3位羟基选择性氧化转换为羰基,得到了一种新型酮内酯塞红霉素(图3),也进入到了临床Ⅲ期研究[17]。
WANG等人将红霉素6位和11位连接为桥环结构后引入了吡啶咪唑环,得到了一系列新型酮内酯,其中抗耐药性最好的化合物EDP-420(modithromycin)(图3)进入到了临床Ⅱ期研究[18]。
3 C-9位引入芳香侧链
红霉素C-9位羰基成肟后,可以提高药物脂溶性和稳定性,同时肟羟基也可以成为新修饰位点,可连接烯丙基或炔丙基进而偶联一系列芳杂环等。
LIANG课题组开发了结构新颖的9-肟酮内酯衍生物,其C-9位引入的烯丙基和炔丙基末端连接的喹啉基和异喹啉杂环化合物(图4)极大提高了抗耐药菌活性,其对革兰氏阳性菌的抗菌活性与泰利霉素相当,高于克拉霉素[19-20]。但不足的是其抗革兰氏阴性菌活性至少8~16倍低于克拉霉素和泰利霉素[21]。除肺炎链球菌、金黄色葡萄球菌和化脓链球菌外,卡他莫拉菌和嗜血流感杆菌也是社区获得性细菌性肺炎的重要病原菌。
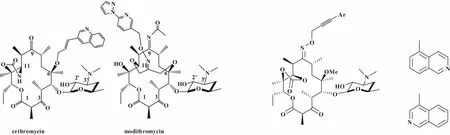
图3 cethromycin and modithromycin 图4 Structure of compounds 9-oxime ketolides
该课题组又合成了一系列C-9位引入含氨基芳环的酮内酯衍生物(图5),活性测试结果表明其对革兰氏阳性菌如肺炎链球菌和化脓性链球菌的抑菌活性和杀菌活性与克拉霉素、泰利霉素相当。更重要的是,其对革兰氏阴性菌如卡他莫拉菌和嗜血流感菌的抗菌活性也取得了突破,可能因芳基上的氨基与靶点形成了氢键作用力大大提高了抗菌活性,解决了之前抗革兰氏阴性菌活性差的难题,为进一步突破对革兰氏阴性菌具有高活性的酮内酯结构修饰方向奠定了重要基础[22]。
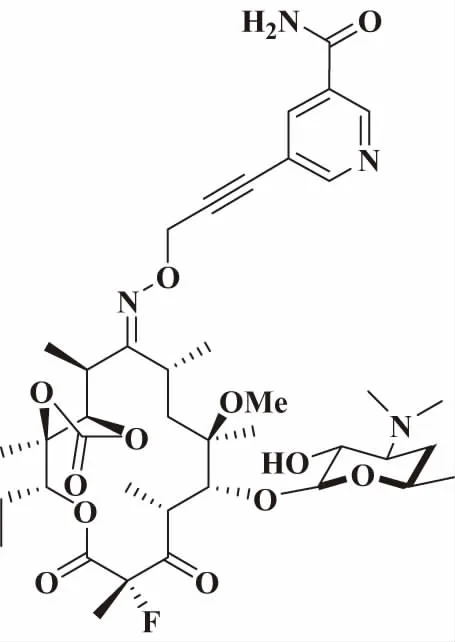
图5 Structure of novel ketolide
MA等人[23]通过杂合手段引入3-羧基喹诺酮结构,抗组成型耐药菌活性有所提高,首次引入3-氨基甲酰基喹诺酮开发了第四代红霉素,进一步提高了抗组成型耐药菌活性,优于对比类似结构药物如克拉霉素和环丙沙星等,体内药代动力学研究显示,该类化合物体内代谢稳定性高于泰利霉素,可有效避免泰利霉素肝毒性。为今后大环内酯类药物结构修饰研究提供了新思路,大环内酯类药物与其他抗菌素类杂合物可能会为解决抗生素耐药性提供一条新的结构修饰道路[24]。
4 结语及展望
红霉素C-3位的克拉定糖是能否克服耐药性的关键所在,围绕克拉定糖的修饰是第三代红霉素研究的重点。随着分子杂合策略成为药物研发的一种重要手段,该策略近期已被应用到大环内酯类抗生素构效关系研究中,例如有研究将喹诺酮类化合物结合于阿奇霉素C-6位上,发现阿奇霉素与环丙沙星/加替沙星的杂合体可以抑制细菌DNA复制,从而提高化合物对高水平耐药菌的抗菌活性[25]。这种既可抑制蛋白合成又可抑制DNA复制的双重作用模式为未来合理设计抗耐药菌的新型抗生素奠定了基础。实现大环内酯与其他抗菌类结构杂合物的合成,在结构新颖性上会成为另一重大突破,甚至有可能找到全新的抗菌机制,同时也会扩大大环内酯类药物的抗菌谱,为大环内酯类药物结构修饰开创新方向与思路。
猜你喜欢
中国药物滥用防治杂志(2022年7期)2022-08-11
山东农业大学学报(自然科学版)(2021年3期)2021-07-29
世界农药(2019年2期)2019-07-13
水运管理(2018年5期)2018-08-20
中国实用医药(2016年30期)2016-12-28
中国实用医药(2016年22期)2016-08-19
中国民族民间医药·下半月(2011年10期)2011-12-27
