
Nicotine-induced adrenal beta-arrestin1 upregulation mediates tobacco-related hyperaldosteronism leading to cardiac dysfunction
2020-06-12 08:57:00NatalieCoraJenniferGhandourCelinaMariePollardVictoriaLynnDesimineKrystenElaineFerrainoJanelleMariePereyraRachelValienteAnastasiosLymperopoulos
World Journal of Cardiology 2020年5期
Natalie Cora, Jennifer Ghandour, Celina Marie Pollard, Victoria Lynn Desimine, Krysten Elaine Ferraino,Janelle Marie Pereyra, Rachel Valiente, Anastasios Lymperopoulos
Abstract
Key words: Adrenocortical zona glomerulosa cell; Aldosterone; βarrestin; Nicotine;Signal transduction; Tobacco-related heart disease
INTRODUCTION
Aldosterone exerts various deleterious effects on the failing heart, while elevated in chronic heart failure (HF)[1-4]. Accordingly, aldosterone levels serve as biomarker of HF severity[5]and mineralocorticoid receptor antagonists have several beneficial effects in HF[6,7]. Aldosterone is produced upon renin-angiotensin-aldosterone-system(RAAS) activation[8]. Together with Angiotensin II (AngII), it exerts a variety of cardiovascular effects in order to maintain renal perfusion and correct electrolyte/blood volume imbalances[1]. In the presence of heart disease however,aldosterone is markedly elevated, hindering cardiac function[1-4].
The main compound in tobacco, nicotine, and its major metabolite in humans,cotinine[9], have been reported to initially inhibit adrenal aldosterone production,causing compensatory RAAS activation upon chronic use in humans (i.e., in chronic smokers)[10-14]. This chronic RAAS activation leads to chronic elevation of aldosterone levels in smokers[10-14]. Given the harmful effects of both AngII and aldosterone in the heart and blood vessels, RAAS activation contributes to HF development in chronic tobacco smokers.
Aldosterone is produced by adrenocortical zona glomerulosa (AZG) cells in response to AngII acting through its type 1 receptors (AT1Rs)[8,15,16]. AT1Rs are G protein-coupled receptors[8]that can also signal through G protein-independent pathways[17-19]. The two universal receptor adaptor proteins β arrestin-1 and -2 (also known as arrestin-2 and -3, respectively) play a central role in mediating this G protein-independent signaling[17,18]. AngII stimulates aldosterone productionviaGq/11-mediated activation of the extracellular signal-regulated kinase (ERK)1/2[20]. ERKs upregulate Steroidogenic Acute Regulatory (StAR) protein, which increases mitochondrial uptake of cholesterol to initiate steroid biosynthesis[15,21,22]. βarrestin1 is a crucial mediator of AT1R signaling to aldosterone production and secretion from human AZG cells[22-29]. The molecular signaling mechanism underlying this crucial role of βarrestin1 in adrenal aldosterone production also involves activation of ERK1/2, which upregulate StAR and, ultimately, aldosterone synthesis and release[22,28].
Since nicotine and cotinine activate RAAS and promote AngII actions at its various tissue targets, including aldosterone production in the adrenal cortex, we hypothesized that these tobacco compounds may chronically increase AngIIdependent aldosterone production in AZG cells, possiblyviaadrenal βarrestin1 upregulation. Indeed, we found that this is the case both in AZG cellsin vitroandin vivo.
MATERIALS AND METHODS
Materials
All chemicals (nicotine, cotinine, AngII, DMSO) were from Sigma-Aldrich (St. Louis,MO, United States; ≥ 98% purity, as assessed by High Performance Liquid Chromatography).
H295R cell culture and transfections
H295R cells were purchased from American Type Culture Collection (Manassas, VA,United States; RRID: CVCL_0458) and cultured, as previously described[22,26]. For siRNA-mediated knockdown, cells were transfectedviathe Lipofectamine method(Invitrogen, Carlsbad, CA, United States) with a custom-ordered rat βarrestin1(Arrb1)-specific siRNA or control scrambled siRNA constructs (custom-made by Sirion Biotech, Cambridge, MA, United States). 48 h after transfection, cells were placed in serum-free medium and treated with the indicated agents for the indicated times.
Aldosterone measurements
In vitro aldosterone secretion in the culture medium of H295R cells and aldosterone levels in rat blood serum were measured by EIA (Aldosterone EIA kit, Cat. #: 11-AD2HU-E01; ALPCO Diagnostics, Salem, NH, United States), as described[22-26].
Real-time polymerase chain reaction
Total RNA isolation with TRIzol reagent (Life Technologies, Grand Island, NY,United States), reverse transcription and real-time polymerase chain reaction (RTPCR) were carried out as previously described[30-32]. The following primer pairs were used: 5′GGCCCCGAGACTTCGTAA3′ and 5′TGGCAGCCACCCCTTGA3′ for rat StAR; 5′CCACATCGGGAAGTTCCAGA-3′ and 5′-CAGGCCGCTGACGAGCAA-3′for rat βarrestin1; 5′-TCAAGAACGAAAGTCGGAGG-3′ and 5′-GGA CAT CTA AGGGCATCAC-3′ for 18S rRNA. Real time PCR was performed using SYBR®Green Supermix (Bio-Rad Laboratories, Hercules, CA, United States). Normalization was done with the housekeeping gene 18S rRNA levels. No bands were seen in the absence of reverse transcriptase.
Western blotting
H295R cell and rat adrenal protein extracts were prepared as described previously[22,23], in a 20 mmol/L Tris pH 7.4 buffer containing 1% Nonidet P-40, 20%glycerol, 10 mmol/L PMSF, 1 mmol/L Na3VO4, 10 mmol/L NaF, 2.5 µg/mL aprotinin, and 2.5 µg/mL leupeptin. Protein concentration was determinedviathe BCA method and equal amounts of protein per sample were loaded. The following antibodies were used for immunoblotting: sc-28869 (Santa Cruz Biotechnology, Santa Cruz, CA, United States) for βarrestin1; sc-25806 (Santa Cruz Biotechnology) for StAR;and sc-47724 (Santa Cruz Biotechnology) for GAPDH. Immunoblots were revealed by enhanced chemiluminescence (ECL, Life Technologies, Grand Island, NY, United States) and visualized in the FluorChem E Digital Darkroom (Protein Simple, San Jose, CA, United States), as described previously[23-26]. Densitometry was performed with the AlphaView software (Protein Simple) in the linear range of signal detection(on non-saturated bands).
Experimental animals and adrenal-specific siRNA delivery
All animal procedures and experiments were performed in accordance with the guidelines of the IACUC committee of Nova Southeastern University. Adrenalspecificin vivosiRNA delivery in -300 g adult (3-month-old) male Sprague-Dawley rats was done, essentially as described[23,30,33],viadirect injection of 1 μg total siRNA[dissolved in sterile phosphate-buffered saline], in each of the two adrenal glands of each animal with a 31-gauge needle. Daily i.p. injections of 1 mg/kg nicotine (or saline), starting on the day of the adrenal-specific siRNA delivery, followed for 7 d in a row. Groups of five animals per treatment were generally used for analysis.
Echocardiography
Two-dimensional gui ded M-mode and Doppler echocardiography using a 14-MHz transducer (Vevo 1100 Echograph, FUJIFILM Visualsonics, Inc., Toronto, ON,Canada) were performed in rats, as described previously[23,25,30]. Three independent echocardiographic measurements were taken in both modes. Echocardiography was performed immediately prior to the adrenal siRNAin vivodeliveries and then again at the end of the nicotine (or saline) treatments. The operator was blind regarding the type of treatment (Arrb1or scrambled siRNA and drug or saline) each animal that was echo'd had received.
Statistical analyses
Data are generally expressed as mean ± SEM. Unpaired 2-tailed Student'sttest and one- or two-way ANOVA with Bonferroni or Dunnett's test was performed for statistical comparisons using the SPSS 23 software (SPSS, Inc., Chicago, IL, United States). For all tests, aP< 0.05 was generally considered to be significant. All sample sizes were calculated for a one-way ANOVA with equal sample sizes in each group and based on previous publications and preliminary data. For the animal experiments, estimation of sample size was done using nQuery Advisor 7.0 software(Informer Technologies, Inc.).
RESULTS
Tobacco compounds upregulate βarrestin1 in AZG cells
To determine whether βarrestin1 is involved in tobacco-dependent adrenal aldosterone production, we took advantage of the human AZG cell line H295R, which endogenously expresses the AT1R (but not the AT2R) and βarrestin1[15,22]. This cell line produces and secretes aldosterone in response to AngII stimulation[15]. Treatment of H295R cells with standard concentrations (10 μM) of either nicotine or cotinine (10µmol/L is very close to the cotinine concentration attained in chronic smokers[9]) for 24 h led to significant upregulation of both mRNA (Figure 1A) and protein (Figure 1B and C) levels of βarrestin1.
βarrestin1 mediates tobacco-induced enhancement of AngII-dependent aldosterone production in AZG cells
Given that βarrestin1 is critically involved in AngII-dependent aldosterone production in AZG cells[34,35], we next examined the impact of its tobacco-induced upregulation on aldosterone turnover in H295R cells. As expected, both nicotine and cotinine markedly enhanced AngII-induced aldosterone secretion (Figure 2A) from H295R cells, as well as StAR mRNA (Figure 2B) and protein (Figure 2C and D) levels in these cells.
Since StAR upregulation signals aldosterone biosynthesis, these results suggest that tobacco compounds significantly enhance aldosterone synthesis and secretion in AZG cells. To prove that this augmentation of aldosterone production by nicotine/cotinine is mediated by the tobacco-upregulated βarrestin1, we knocked it downviasiRNA in H295R cells (Figure 3A) and treated them again with nicotine and cotinine to examine the effect on aldosterone synthesis and secretion. Indeed, βarrestin1 knockdown almost completely abrogated the nicotine- and cotinine-induced enhancement of AngII-dependent aldosterone secretion (Figure 3B), StAR mRNA levels (Figure 3C),and StAR protein levels (Figure 3D and E) in H295R cells.
Adrenal βarrestin1 siRNA-mediated knockdown ameliorates tobacco-induced hyperaldosteronism and cardiac dysfunction in vivo
To demonstrate the physiological significance of ourin vitrofindings in H295R cells,we also treated adult male (otherwise healthy) rats with daily i.p. injections of nicotine for 7 d and, at the end of the 7-d-long treatment period, we measured their circulating aldosterone levels and cardiac function [ejection fraction (EF %)]. Of note,the daily dose of nicotine administered (1 mg/kg per day) is known to lead to blood circulating levels of nicotine comparable to those of chronic human smokers[9]. As shown in Figure 4A, nicotine exposure led to a significant hyperaldosteronism in these animals after one week of treatment. This led to cardiac dysfunction beginning to set in, since the nicotine-exposed animals displayed slightly but, nevertheless,significantly less EF compared to control saline-injected animals (Figure 4B). Notably,all animals used in the study had comparable EF at the beginning of the 7-d-long treatments (i.e., prior to group randomization) (69% ± 4.2%).
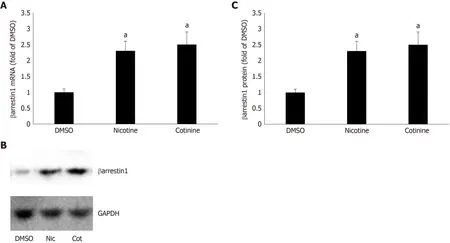
Figure 1 Effect of tobacco compounds on βarrestin1 levels in AZG cells. A: βarrestin1 mRNA levels in H295R cells treated for 24 h with 10 μmol/L nicotine or 10 μmol/L cotinine or vehicle. B-C: Protein levels in H295R cells treated for 24 h with 10 μmol/L nicotine or 10 μmol/L cotinine or vehicle. Representative western blots are shown in (B), along with glyceraldehyde 3-phosphate dehydrogenase as loading control, and the densitometric quantitation of three independent cell extracts per condition run in duplicate (and normalized with glyceraldehyde 3-phosphate dehydrogenase levels) is shown in (C). aP < 0.05 vs vehicle, n = 3 independent experiments/treatment in duplicate. DMSO: Vehicle; Cot: Cotinine; Nic: Nicotine; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase.
Importantly, in rats having βarrestin1 knocked-downviasiRNA specifically in their adrenals (Figure 4C), nicotine exposure at the same concentration and for the same time-period (7 d) led to substantially less hyperaldosteronism (Figure 4D) and better cardiac function (higher EF) (Figure 4E) than in control animals receiving scrambled siRNA in their adrenals at the end of the 7-day-long treatments. Of note, adrenal βarrestin1 knockdown significantly lowered circulating aldosterone levels even in saline-treated animals (Figure 4D), which is consistent with βarrestin1's essential role in adrenal aldosterone productionin vivo[24]. However, this did not translate into better cardiac function of saline-treated animals (Figure 4E), probably because these animals were overall healthy and their cardiac function was optimal to begin with (there was a non-statistically significant trend toward higher EF though also in the saline-treated,Arrb1-siRNA rats, see Figure 4E). In any case, taken together, thein vivoresults of Figure 4 strongly suggest that adrenal βarrestin1 mediates tobacco-related hyperaldosteronism and cardiac dysfunctionin vivo, as well.
DISCUSSION
In the present study, we have identified adrenal βarrestin1 as a novel molecular target for mitigating the aldosterone-dependent cardiotoxic effects of tobacco. AngII induces aldosterone production in AZG cells by binding to its adrenal AT1R, which then activates βarrestin1[22]. Nicotine and cotinine are known to activate RAAS[13],promoting hyperaldosteronism. We report herein that these tobacco compounds chronically upregulate adrenal βarrestin1, promoting excessive aldosterone synthesis and secretion from human AZG cells in vitro and from adrenal glandsin vivo. Thus,adrenal βarrestin1 appears to be a crucial component of tobacco-induced RAAS activation, which contributes to heart disease development/progression.
AngII-dependent aldosterone secretion and aldosterone biosynthesis, as measured by the expression levels of StAR, the rate-limiting enzyme in aldosterone biosynthesis,were found significantly higher in H295R cells treated with either nicotine or cotinine compared to control, vehicle-treated cells. Importantly, we uncovered that this was due to significant upregulation of βarrestin1, at both the mRNA and protein levels, in these cells, since βarrestin1 siRNA-mediated knockdown reversed the tobacco compound-induced increases in AngII-dependent aldosterone secretion and in StAR expression (i.e. aldosterone biosynthesis).
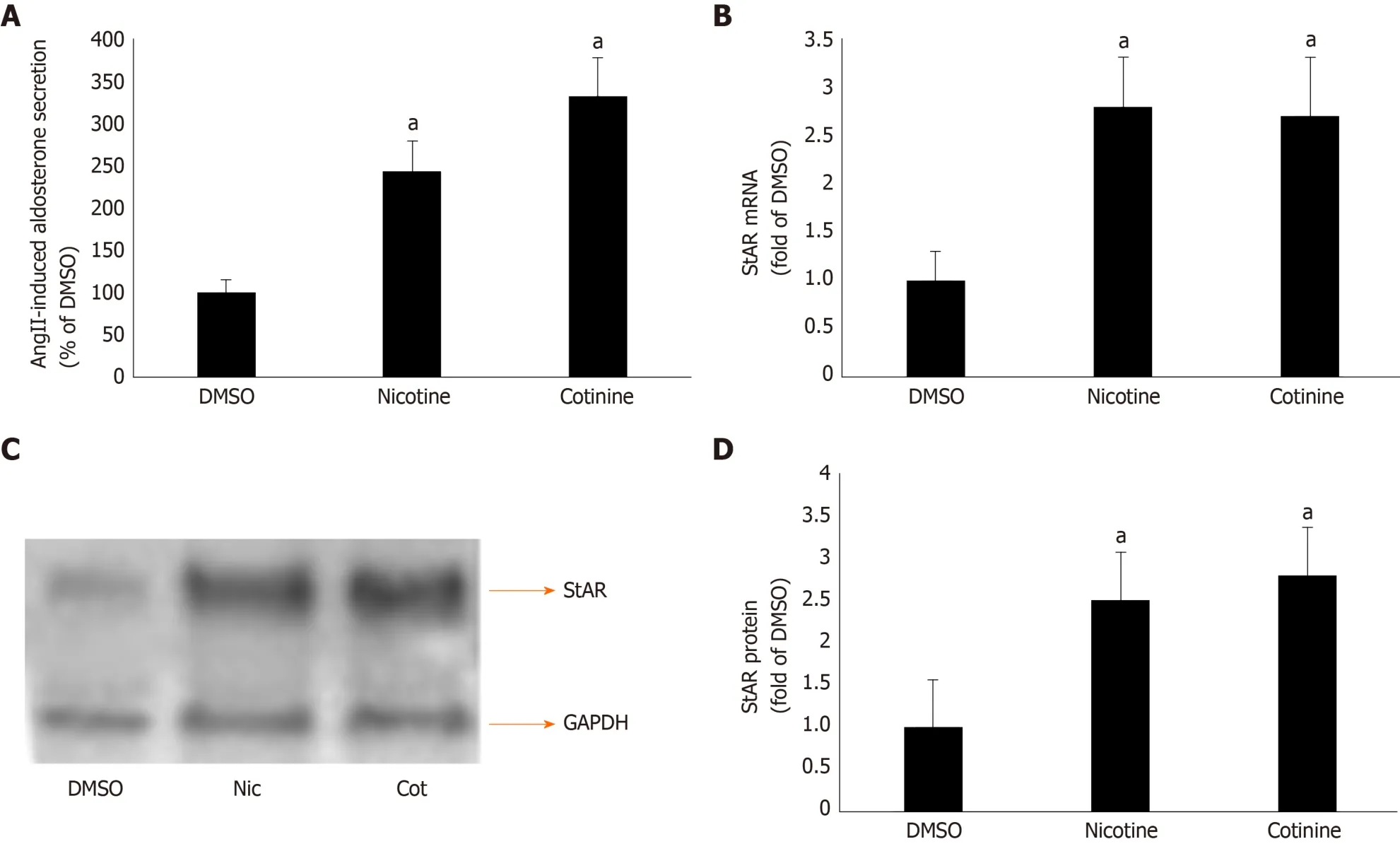
Figure 2 Effect of tobacco compounds on AngII-dependent aldosterone synthesis and secretion in adrenocortical zona glomerulosa cells. A: Aldosterone secretion into the culture medium from H295R cells in response to a 6-hour-long 100 mmol/L AngII challenge, as measured 24 h post-treatment with 10 μmol/L nicotine or 10 μmol/L cotinine or vehicle (DMSO). Data are expressed as % of the AngII response in DMSO-treated cells. aP < 0.05, vs DMSO; n = 5 independent measurements per treatment performed in duplicate; B: StAR mRNA levels in H295R cells treated for 24 h with 10 μmol/L nicotine (Nic) or 10 μmol/L cotinine (Cot) or vehicle (DMSO). C-D: Protein levels in H295R cells treated for 24 h with 10 mol/L nicotine (Nic) or 10 μmol/L cotinine or DMSO. Representative western blots are shown in (C), along with glyceraldehyde 3-phosphate dehydrogenase as loading control, and the densitometric quantitation of three independent cell extracts per condition run in duplicate (and normalized to glyceraldehyde 3-phosphate dehydrogenase) is shown in (D). aP < 0.05, vs DMSO; n = 3 independent experiments/treatment in duplicate. DMSO: Vehicle; Cot: Cotinine; Nic: Nicotine; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase.
Of note, chronic nicotine exposure increased adrenal βarrestin1-mediated aldosterone synthesis and secretion, causing hyperaldosteronism, alsoin vivo. This led to development of cardiac dysfunction (ejection fraction drop/functional decline) in animalsin vivo. As a proof of concept, adrenal-specific βarrestin1 siRNA-mediated knockdownin vivonormalized the elevated circulating aldosterone levels and significantly attenuated the cardiac functional decline induced by the chronic nicotine exposurein vivo. These findings strongly suggest that adrenal βarrestin1 inhibition (or genetic knockdown/ablation) might be of value in prevention or amelioration of tobacco-related heart disease progression and risk elevation.
The mineralocorticoid receptor (MR) is known to underlie HF pathology and it was recently documented to promote cardiac dysfunction and cardiomyopathy in transgenic mice, even in the absence of a cardiac insult[36]. Thus, aldosterone, the endogenous natural MR agonist, needs to be suppressed for heart disease therapy.Given various reports that this hormone oftentimes acts in an MR-independent manner[37], which circumvents the actions of MR antagonist (MRA) drugs, cutting aldosterone production at its source,i.e. the adrenal cortex,viaβarrestin1 inhibition poses as an even more efficient approach to combat aldosterone's (and tobacco's)cardiotoxic actions than simply using MRA's.
Adrenal βarrestin1 blockade has been shown to effectively suppress adrenal aldosterone production[28]. This is obviously feasibleviagene therapy to knock down or knock out the protein specifically from the adrenal glands. Pharmacologic blockade of the adrenal AT1R with candesartan or valsartan, which are very potent βarrestin1 inhibitors, is another possible approach[25,26]. Of course, whether any ARB, like candesartan or valsartan, can effectively suppress the nicotine (tobacco)-induced elevation in aldosterone production from AZG cells is an open question right now and one that needs to be addressed in future studies. Alternatively, the βarrestin1-mediated signaling to aldosterone synthesis could be targeted with barbadin, a compound that was recently identified as an inhibitor of βarrestin-dependent internalization and signaling[38].
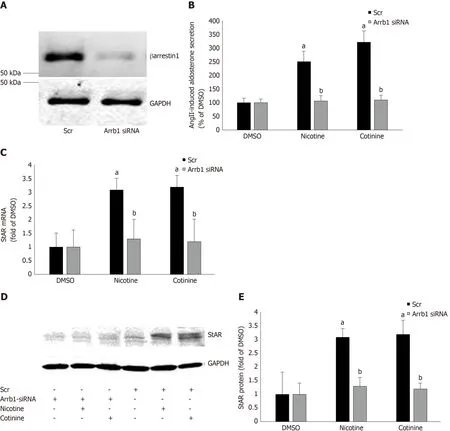
Figure 3 Effect of βarrestin1 knockdown on tobacco-induced aldosterone production in adrenocortical zona glomerulosa cells in vitro. A: Immunoblotting for βarrestin1 in H295R cell extracts 48 h post-transfection with βarrestin1-sepcific (Arrb1) siRNA or scrambled (Scr) siRNA to test the efficiency of the βarrestin1 siRNA-mediated knockdown. A representative blot of three independent cell extracts run in duplicate is shown, along with glyceraldehyde 3-phosphate dehydrogenase(GAPDH) as loading control, confirming an > 80% βarrestin1 protein knockdown; B: Aldosterone secretion into the culture medium from H295R cells in response to a 6-hour-long 100 nmol/L AngII challenge, as measured 24 h post-treatment with 10 µmol/L nicotine or 10µmol/L cotinine or vehicle (DMSO) in H295R cells having Arrb1 siRNA or not (scrambled siRNA-transfected, Scr). Drug treatments were carried out 48 h after siRNA transfection (i.e., AngII was added at 72 h posttransfection). Data are expressed as % of the AngII response in DMSO-treated cells. aP < 0.05, vs DMSO (Arrb1 siRNA or Scr); bP < 0.05, vs Scr; n = 5 independent measurements per treatment performed in duplicate; C: StAR mRNA levels in these cells; D-E: Protein levels in these cells. For StAR immunoblotting, representative blots are shown in (D), along with GAPDH as loading control, and the densitometric quantitation of three independent cell extracts per treatment condition run in duplicate (and normalized to GAPDH) is shown in (E). aP < 0.05, vs DMSO (vehicle); bP < 0.05, vs Scr; n = 3 independent experiments/treatment in duplicate. DMSO:Vehicle; Scr: Scrambled; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; Arrb1: Arrestin1-sepcific.
Finally, nicotine (and tobacco in general) has been known for decades to cause sympathetic activation and to increase circulating catecholamines,e.g., by stimulating catecholamine secretion from the adrenal medullaviadirect agonism of nicotinic cholinergic receptors expressed on chromaffin cell membranes[30,39,40]. We report here that it can also promote production and secretion of another major adrenal hormone with important effects on the myocardium and the vasculature,i.e., aldosterone from the adrenal cortex. Nicotine achieves this thanks to direct upregulation of βarrestin1 in AZG cells, an AT1R-adapter protein that is an essential transducer linking the AngII hormone signal with aldosterone production in these cells[28]. Therefore, βarrestin1 might be a crucial molecular master-switch controlling tobacco-dependent stimulation of hormone production in the adrenal gland, which has enormous repercussions for cardiovascular homeostasis, in general, and for the function of the myocardium.
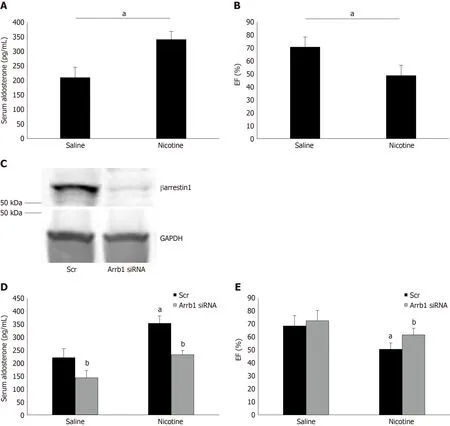
Figure 4 Effect of adrenal βarrestin1 knockdown on nicotine-dependent hyperaldosteronism and cardiac dysfunction in vivo. A: Circulating aldosterone levels in rats injected i.p. with 1 mg/kg per day nicotine or saline (control) for 7 consecutive d. aP < 0.05; n = 5 rats/group; B: Ejection fraction [EF (%)] of these animals at the end of the saline or nicotine treatments. aP < 0.05; n = 5 rats/group; C: Immunoblotting for βarrestin1 in adrenal protein extracts isolated from rats at 7 d postinjection with βarrestin1-specific siRNA or scrambled (Scr) siRNA directly into their adrenal glands. A representative blot of three independent rat adrenal protein extracts is shown, along with glyceraldehyde 3-phosphate dehydrogenase as loading control, confirming an > 90% βarrestin1 protein knockdown in the adrenal glands in vivo; D: Circulating aldosterone levels of rats having adrenal βarrestin1 knocked-down (rrestin1-specific siRNA) or not (scrambled siRNA-injected, Scr) and treated with 1 mg/ kg per day i.p. nicotine or saline (control) for 7 consecutive d, at the end of these treatments. E: EF (%) of rats having adrenal βarrestin1 knocked-down(rrestin1-specific siRNA) or not (scrambled siRNA-injected, Scr) and treated with 1 mg/kg per day i.p. nicotine or saline (control) for 7 consecutive d, at the end of these treatments. aP < 0.05 vs Saline; bP < 0.05 vs Scr; n = 5 rats/group. Scr: Scrambled; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; Arrb1: Arrestin1-sepcific; EF (%): Ejection fraction.
The present study has two major limitations: (1) The small animal group sizes-more data are needed in more animals and with other nicotine doses and routes of administration to fully confirm the present results; and (2) The study has to be repeated in larger animals (e.g., pigs or rabbits) that more closely resemble human physiology and disease conditions. In addition, all of the animals used in the present study were male; experiments need to be repeated in female rats, as well. Finally,AngII is only one of several stimuli for adrenal nicotine-induced aldosterone production. Perhaps different results will be obtained, if the aldosterone response to a different hormone is studied.
In conclusion, nicotine, and its major metabolite in humans cotinine, induce hyperaldosteronismviaadrenal βarrestin1 upregulation, which mediates enhanced AngII-dependent aldosterone production from the adrenal cortex in vitro and in vivo.Thus, adrenal βarrestin1 upregulation is an essential biological mechanism underlying the tobacco-induced increase in RAAS activity observed in chronic smokers and in chronically tobacco-exposed animals. This results in raised aldosterone levels and thus, in increased cardiac dysfunction and cardiovascular risk.Therefore, adrenal βarrestin1 inhibition, either pharmacologically or genetically (viasiRNA-mediated knockdown or even CRISPR/Cas9-mediated gene excision), poses as an attractive therapeutic or even preventive strategy for mitigating the devastatingly toxic effects of tobacco on the heart, by reducing the neurohormonal(aldosterone) burden of the myocardium.
ARTICLE HIGHLIGHTS
Research background
Nicotine, the main addictive compound in tobacco, is associated with major cardiovascular adverse events, such as heart failure and hypertension. One of the molecular mechanisms underlying nicotine-induced cardiotoxicity is elevation of renin-angiotensin-aldosterone system(RAAS) activity. Nicotine, and its major metabolite in humans cotinine, have been reported to induce RAAS activation, resulting in hyperaldosteronism. Aldosterone has myriad adverse cardiac effects and is produced by the adrenal cortex in response to angiotensin II (AngII) acting through its type 1 receptors (AT1Rs). AT1Rs induce aldosterone productionviaboth Gq/11proteins and βarrestin1 (Arrestin-2).
Research motivation
It was hypothesized that nicotine activates adrenal ßarrestin1, thereby contributing to RAAS activation and heart disease development.
Research objectives
We tested our hypothesis by investigating the effects of nicotine on aldosterone production invitroand on aldosterone levels and cardiac function of experimental animalsin vivo.
Research methods
We used the human adrenocortical zona glomerulosa (AZG) cell line H295R, in which we performed real-time polymerase chain reaction (PCR) and western blotting to measure βarrestin1 mRNA and protein levels, respectively, as well as ELISA to measure aldosterone secretion. We also manipulated βarrestin1 expressionviasiRNA-mediated knockdown in H295R cells. For the in vivo studies, we used adult male Sprague-Dawley rats, which we exposed to chronic nicotine administration after adrenal-specific, βarrestin1 siRNA-mediated knockdown or control scrambled siRNA deliveryin vivo.
Research results
Nicotine and cotinine upregulate βarrestin1 mRNA and protein levels in AZG cells, which augments aldosterone synthesis and secretion. In contrast, siRNA-mediated βarrestin1 knockdown mitigates the effects of nicotine on AngII-induced aldosterone production.In vivo,nicotine-exposed experimental rats with adrenal-specific βarrestin1 knockdown display lower circulating aldosterone levels and better cardiac function than nicotine-exposed control animals with normal adrenal βarrestin1 expression.
Research conclusions
Adrenal βarrestin1 upregulation is one of the mechanisms by which tobacco,i.e. nicotine,promotes cardio-toxic hyperaldosteronism that accelerates cardiac functional decline, bothin vitroandin vivo.
Research perspectives
Adrenal βarrestin1 pharmacological blockade or genetic deletion (or knockdown) represents a novel therapeutic strategy to ameliorate tobacco-related heart disease morbidity and mortality.
ACKNOWLEDGEMENTS
The authors would like to acknowledge Dr. Lina Shehadeh, Univ. of Miami Miller School of Medicine (Miami, FL, United States) and members of her laboratory for excellent technical assistance.
 World Journal of Cardiology2020年5期
World Journal of Cardiology2020年5期
- World Journal of Cardiology的其它文章
- Management of hypertension in COVID-19
- Incidental discovery of right ventricular lipoma in a young female aImaging investigations and diagnosissociated with ventricular hyperexcitability: An imaging multimodality approach
- Preoperative nuclear stress testing in the very old patient population
- Access to smart devices and utilization of online health resources among older cardiac rehabilitation participants
- Management of adults with coarctation of aorta
- New guidelines for the diagnosis and management of pulmonary embolism: Key changes