
Isolated colonic neurofibroma in the setting of Lynch syndrome: A case report and review of literature
2020-06-11 04:22WarrenYLSunArmaanPandeyMarkLeeShawnWasilenkoShahzeerKarmali
Warren YL Sun, Armaan Pandey, Mark Lee, Shawn Wasilenko, Shahzeer Karmali
Abstract
Key words: Isolated gastrointestinal neurofibroma; Colonic neurofibroma; Gastrointestinal neurofibromatosis; Lynch syndrome; Case report
INTRODUCTION
Lynch syndrome (LS), or hereditary nonpolyposis colorectal cancer (HNPCC), is an autosomal dominant disorder caused by a germline mutation in one of the DNA mismatch repair genes (MLH1, MSH2, MSH6, or PMS2) that predisposes patients to various malignancies, of which colorectal cancer (CRC) is the most common[1]. Given the increased risk, patients diagnosed with LS are recommended to undergo cancer screening, including screening colonoscopy for CRC every two years beginning at the age of 20 to 25[2]. LS has also been associated with an increased risk of endometrial,gastric, ovarian, hepatobiliary, urinary tract, small bowel, and other cancers[1].Additionally, the association of LS with neurofibromatosis has been reported in literature[3].
Neurofibromas are benign nerve sheath tumours originating from the peripheral nervous system containing an amalgamation of Schwann cells and fibroblasts[4]. While rare, malignant transformation into neurofibrosarcoma, or malignant peripheral nerve sheath tumour (MPNST) can occur[5]. Neurofibromas are typically associated with neurofibromatosis Type 1 (NF1), also known as von Recklinghausen disease. NF1 is an autosomal dominant disorder due to a mutation in chromosome 17[4]. Classic features of NF1 include café-au-lait macules, Lisch nodules, and neurofibromas of the skin; however, the cardiovascular system, eyes, bones, and gastrointestinal system can also be affected[6].
In patients with NF1 or multiple endocrine neoplasia type 2B (MEN 2B),gastrointestinal neurofibromas is relatively common, affecting approximately 25% of patients[7]. Most gastrointestinal neurofibromas occur in the stomach and small intestine[8]. These manifestations are typically asymptomatic, but symptoms may include abdominal pain, gastrointestinal bleeding, obstruction, and palpable masses[6].However, isolated gastrointestinal neurofibromas in patients without a history of NF1 or MEN 2B are extremely rare[7]. In this report, we will present a rare case of isolated colonic neurofibroma in a patient with a history of LS.
CASE PRESENTATION
Chief complaints
A 33-year-old female with LS diagnosed with positive MSH6 mutation presented for routine screening gastroscopy and colonoscopy.
History of presenting illness
The patient was asymptomatic with no gastrointestinal issues or stigmata of neurofibromatosis.
Personal and family history
The patient had a family history significant for LS and colorectal cancer in multiple family members. The patient was also diagnosed with LS with positive MSH6 mutation. She was otherwise healthy. There was no family history of NF1.
Physical examination
Physical examination revealed a benign abdomen. Cutaneous examinations revealed no evidence of café-au-lait spots or neurofibromatosis.
Laboratory examinations
Complete blood count, comprehensive metabolic panel, liver panel, and coagulation studies were all within normal limits. The patient also had a normal carcinoembryonic antigen (CEA) level of 1.3 μg/L (normal range < 5.0 μg/L).
Imaging examinations
Initial screening gastroscopy was unremarkable. Random biopsies revealed no diagnostic abnormality. Initial screening colonoscopy demonstrated 2 to 2.5 cm of abnormal appearing mucosa in the ascending colon with central tethering. An attempt to lift the area of concern with methylene blue was unsuccessful. The area was tattooed and biopsied, which was non-diagnostic. The patient underwent a repeat colonoscopy after two months. The suspicious area was again appreciated appearing as sessile, serrated, polypoid tissue measuring approximately 2.5 cm wrapped around a haustral fold in the ascending colon (Figure 1). Biopsies were obtained again, but the polyp was unresectable. The biopsies revealed sessile serrated polyp, negative for dysplasia. A staging workup was completed, which included a negative computerized tomography (CT) scan of the chest, abdomen and pelvis.
TREATMENT
Following two non-diagnostic biopsies, a referral to general surgery was made for the management of the unresectable polyp in the setting of LS. A discussion with the patient regarding the management included the recommendation of a subtotal colectomy versus a segmental resection, for which the patient elected for the latter.The patient underwent a laparoscopic right hemicolectomy.
FINAL DIAGNOSIS
Gross pathological examination revealed a 2.2 cm × 1.8 cm × 0.4 cm ill-defined polypoid lesion adjacent to the cecum. Microscopic examination demonstrated an area of reactive serrated mucosa overlying an ill-defined submucosal mass extending into superficial muscularis propria and overlying mucosa. The mass consisted of short small monomorphic spindle cells within a collagenous background (Figure 2A). The spindle cells were intermixed with scattered mast cells (Figure 2B).Immunohistochemistry showed the spindle cells were positive for S100 (Figure 2C).Fifteen lymph nodes were identified and negative for malignancy. Overall, the histomorphology and phenotype by ancillary immunohistochemistry were consistent with a gastrointestinal neurofibroma.
OUTCOME AND FOLLOW-UP
The patient tolerated the procedure well and was discharged on postoperative day three. She was seen one month later in follow-up and had recovered well. She will not require any further treatment with regards to her isolated colonic neurofibroma, but will continue to follow-up for ongoing surveillance of her LS.
DISCUSSION
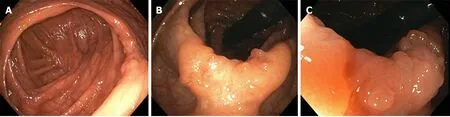
Figure 1 Endoscopic imaging of gastrointestinal neurofibroma. A: A 2.5 cm polypoid lesion was identified within a haustral fold adjacent to the cecum. B: The lesion demonstrated abnormal mucosa with central tethering suspicious for malignancy. C: Magnified view of polypoid lesion with abnormal mucosa concerning for adenocarcinoma.
In this report, we presented the first case of isolated colonic neurofibroma in the setting of LS. Screening colonoscopy is routinely performed in patients with LS to diagnose colorectal cancers at an earlier stage. Commonly, clinicians and patients with non-diagnostic biopsies of suspicious colonic masses are faced with a dilemma: To resect or not resect. Given the patient in this study was at an increased risk of CRC due to LS, aggressive management of the mass with resection is appropriate.However, while submucosal aetiologies are more rare, it is also important to consider them in patients with non-diagnostic biopsies of colonic masses that may appear to have endoscopic features of submucosal origin. When conventional endoscopic mucosal biopsies fail to diagnose masses, the use of endoscopic ultrasound (EUS)guided biopsies may be considered to aid in the diagnosis[9].
Given the clinical context in this study, the patient was presented with the option of segmental resection versus subtotal colectomy. The patient expressed a desire for future pregnancy, and therefore, we elected for segmental resection to reduce the risk of infertility. However, total colectomy with ileoanal anastomosis would be the preferred primary treatment for patients with colon cancer or colon neoplasia unresectable by endoscopy[1]. The patient will require ongoing surveillance for CRC in the remainder of her colon. Furthermore, the patient would have required a completion proctocolectomy if her pathology confirmed a CRC.
Surprisingly, the final pathology revealed a gastrointestinal neurofibroma.Furthermore, the patient did not exhibit any features of NF1, thus, classifying this case as an isolated colonic neurofibroma. There have only been fourteen cases of isolated colorectal neurofibromas reported in the English literature (Table 1). The average age of presentation was 51 years. Nine patients (64%) were female, and four (29%) were asymptomatic.
While LS has not been associated with neurofibromas, MSH6 mutation has been associated with NF1 phenotype in literature[10]. Given that gastrointestinal neurofibromas are rarely found in isolation, some have recommended referring patients with isolated gastrointestinal neurofibromas for the workup and surveillance of NF1[4,8,11-14]. We also agree with the need to screen patients with isolated gastrointestinal neurofibromas for NF1 given the associated morbidity and mortality with these conditions.
CONCLUSION
We present the first case of an isolated colonic neurofibroma in a patient with LS.Given the risk of colorectal cancer, the patient had a non-diagnostic polyp resected.There are currently no guidelines for the management of isolated gastrointestinal neurofibromas due to the lack of studies. We recommend considering establishing a diagnosis with endoscopic mucosal biopsy or ultrasound guided biopsy, reserving resection for patients with symptomatic disease or alternative indications, and continuing to follow patients for the surveillance of NF1 to reduce associated morbidity and mortality.

Figure 2 Pathologic examination of gastrointestinal neurofibroma. A: Low power view of submucosal spindle cell proliferation, (hematoxylin-eosin, 2.5 ×). B:Higher power view of monomorphic spindle cells with intermixed mast cells, (hematoxylin-eosin, 10 ×). C: Immunohistochemistry staining showing variable positivity of lesional cells for S100 protein (10 ×).
 World Journal of Gastrointestinal Surgery2020年1期
World Journal of Gastrointestinal Surgery2020年1期
- World Journal of Gastrointestinal Surgery的其它文章
- Bile leakage after loop closure vs clip closure of the cystic duct during laparoscopic cholecystectomy: A retrospective analysis of a prospective cohort
- Outcomes associated with the intention of loco-regional therapy prior to living donor liver transplantation for hepatocellular carcinoma
- Pathological abnormalities in splenic vasculature in non-cirrhotic portal hypertension: Its relevance in the management of portal hypertension