
探讨PM2.5暴露的肺外毒理效应对绝经后骨代谢的影响机制
2020-06-06 08:40刘宗玉黄惠娟杨帆曾小娟
中国骨质疏松杂志 2020年4期
刘宗玉 黄惠娟 杨帆 曾小娟
厦门大学附属东方医院妇产科,福建 福州 350025
绝经后骨质疏松症(postmenopausal osteoporosis, PMOP)是衰老相关性低雌激素效应,导致高效骨重塑向骨吸收方向进行致骨脆性增加、易于骨折的全身性骨病,是骨质丢失中最常见的一种类型[1]。我国人口基数大,人口老龄化严峻,上海部分地区调查发现,骨质疏松造成髋部骨折中,中老年女性是男性3倍多,人均住院费用高达3万余元,平均住院天数近18 d[2]。骨质疏松的高发病率及其并发症的高致残率、高致死率及高消耗医疗、经济资源问题,使得PMOP成为我国医疗卫生面临的重大挑战。PM2.5易吸附大量氧化还原特性的过渡金属、持久的自由基、多环芳烃(PAHs)等有害物质,并具有极强的穿透力,极易破坏肺泡毛细血管屏障随血液作用于全身脏器,改变机体多种生物活性,包括炎症细胞的招募,释放大量炎性因子和活性氧(ROS)等,对人体毒害最强[3-5]。我国京津冀、长三角,珠三角年平均PM2.5分别为70、72、53μg/m3[6]均高于我国规定的二级年均水平限值35μg/m3,提示我国环境污染严峻。环境污染对破骨/成骨细胞的杀伤作用与氧化应激及活性氧等相关,其可使RANK/RANKL/OPG比值失衡和下调抗氧化酶, 引起骨质丢失[7]。现就氧化应激、炎症因子、雌激素及相关受体(如AhR)等对骨质疏松的影响进行综述。
1 PM2.5暴露的毒性效应与绝经后骨代谢密切相关
1.1氧化应激及活性氧
PM2.5参与机体生物活性转化过程,可诱导炎症和氧化损伤产生具有协同效应的活性氧或氮(如ROS、RNS)和炎症介质[8]。氧化应激及活性氧被广泛认为是许多病理条件下的致病因素,包括骨质疏松。缺氧可使骨髓基质细胞中的线粒体产生大量活性氧,其在低雌激素水平能强有力促进骨吸收[9-11]。此外,雌激素缺乏可致相关组织主要抗氧化剂、抗氧化酶水平显著下降,活性氧(如H2O2)水平升高,降低骨的抗氧化防御能力,运用抗氧化剂可阻止卵巢切除后引起的骨质疏松[11-14]。Altindag等[15]通过测定血浆TAS、TOS水平获得OSI时,发现血浆中TOS和OSI值在绝经后骨质疏松症患者中明显升高,血浆TAS水平明显降低,而腰椎和股骨颈部位骨密度与OSI呈显著负相关。Baek等[16]通过测定血清DNA氧化损伤的标志物8-羟基-2O-脱氧鸟苷(8-OH-DG)水平,同样发现8-OH-DG水平与腰椎、全髋关节、股骨颈和粗隆部的骨密度水平呈负相关。
另外,职业性暴露于低水平PM2.5(12.4±6.9 μg/m3)和BaP(1.0±0.6 ng/m3)的出租车司机血清炎症生物标志物水平明显升高,活性氧和相关氧化损伤标志物增加,抗氧化酶和抗炎介质(IL-10)下降[17]。反过来,升高的活性氧又可增加吸收性炎症因子的表达间接刺激破骨细胞[18-19]。雌激素缺乏致骨髓微环境中负调控炎症因子、氧化物等系统地和局部地增加,并通过TGF-β的表达延缓破骨细胞的凋亡,诱导破骨细胞分化,引发骨质疏松[20]。促炎细胞因子被证明能提高不同类型细胞内的ROS水平[21]。而外源性ROS或内源性ROS均可刺激破骨细胞的形成和吸收活动,但其细节尚不清楚。PM2.5可促进细胞内活性氧生成和NF-κB磷酸化,ROS(主要H2O2)可刺激M-CSF和RANKL的表达,提高RANKL/OPG的比值,使低雌激素水平妇女骨吸收增加和骨量降低[15-16,22]。最新研究指出,RANKL能参与Bach 1的核易位,抑制Nrf 2依赖的抗氧化酶的表达,并通过与Bach 1核蓄积竞争,从而增强破骨细胞内的ROS信号[23]。应用抗氧化剂能减少RANKL诱导的Akt、NF-kB和ERK活化,显著降低RANKL诱导的骨吸收活性和破骨细胞存活率,促进破骨发生[22]。此外,ROS还可通过MAPK和Ca2+介导的信号通路参与骨代谢[21]。
1.2炎症细胞因子
目前国内关于PM2.5诱发促炎因子直接或间接引发绝经后骨质疏松的相关研究并不多见。Zheng等[24]发现绝经后妇女腰椎骨密度与血清炎症细胞因子IL-1、IL-6和TNF-α呈负相关性,提示雌激素可调节各种细胞因子的分泌或释放阻止骨质丢失。Terauchi等[25]指出骨髓中Th细胞分泌的IL-1、IL-6和TNF-α等均可促进骨髓破骨细胞的发生、增殖,引发骨质疏松。PM2.5在肺中的沉积可引起全身炎症反应,并能刺激骨髓产生广谱的促炎细胞因子,且颗粒直径越小,血清中产生的炎症细胞因子越多,引发相应脏器病理损害越严重[26]。研究发现慢性暴露于PM2.5可使血清中的TNF-α、IL-1、IL-6、IFN-γ等炎症生物标记物水平明显升高,导致血清RANKL值增加41%,OPG下降22%,引起该群体骨量减少[17,27-28]。
炎症细胞因子在缺氧、感染和炎症期间对骨破坏发挥着关键作用。雌激素缺乏可通过抗原提呈细胞和细胞因子IFN-γ、IL-7和TGF-β介导的复杂细胞机制诱导系统地和局部地增加炎症细胞因子分泌或释放[25]。强效骨吸收因子(TNF-α、IL-1、IL-6)能通过分布在破骨细胞前体上相应的受体(如TNFR、IL-1 R和IL-6R)发挥其生物学效应,在对其相应受体缺陷小鼠的分析表明,TNFR 1、IL-1 RI、sIL-6R促进破骨细胞分化,而TNFR 2、IL-1 RII抑制破骨细胞分化,TNF-α、IL-1、IL-6能增强TNFR 1、IL-1 RI、sIL-6R破骨发生,降低TNFR 2、IL-1 RII功能,促进破骨发生[29-31]。研究发现敲除TNF-α、IL-1 R、IL-6基因的动物可抑制卵巢切除后骨质丢失,证实细胞因子对绝经后骨代谢的影响是必不可少的、多途径的[32-34]。
最后,炎症介质直接或间接通过激活共同信号通路OPG/RANKL/RANK调节骨代谢。这些细胞因子可提高RANKL mRNA的表达和RANKL/OPG的比值,激活下游信号磷酸化(如JNK、p38、MAPK、JAK2、ERK和Akt)促进成骨细胞介导的破骨细胞分化、成熟;还可直接强烈激活NF-κB、p38、ERK和应激激活蛋白激酶/c-Jun NH2-末端激酶活性,并显著降低ALP活性及成骨细胞基因(RunX 2、Osterix和骨钙素)的表达,且呈剂量依赖性抑制骨矿化和成骨细胞分化,从而导致女性骨质丢失加重[34-38]。炎症因子还能强烈抑制成骨细胞的生成,这种抑制成骨细胞活性的途径是通过影响细胞因子下游信号通路,诸如MAPK、Stats、SMURF 1/2、PGE2等[36,38-43],使新骨形成无法代偿骨吸收。
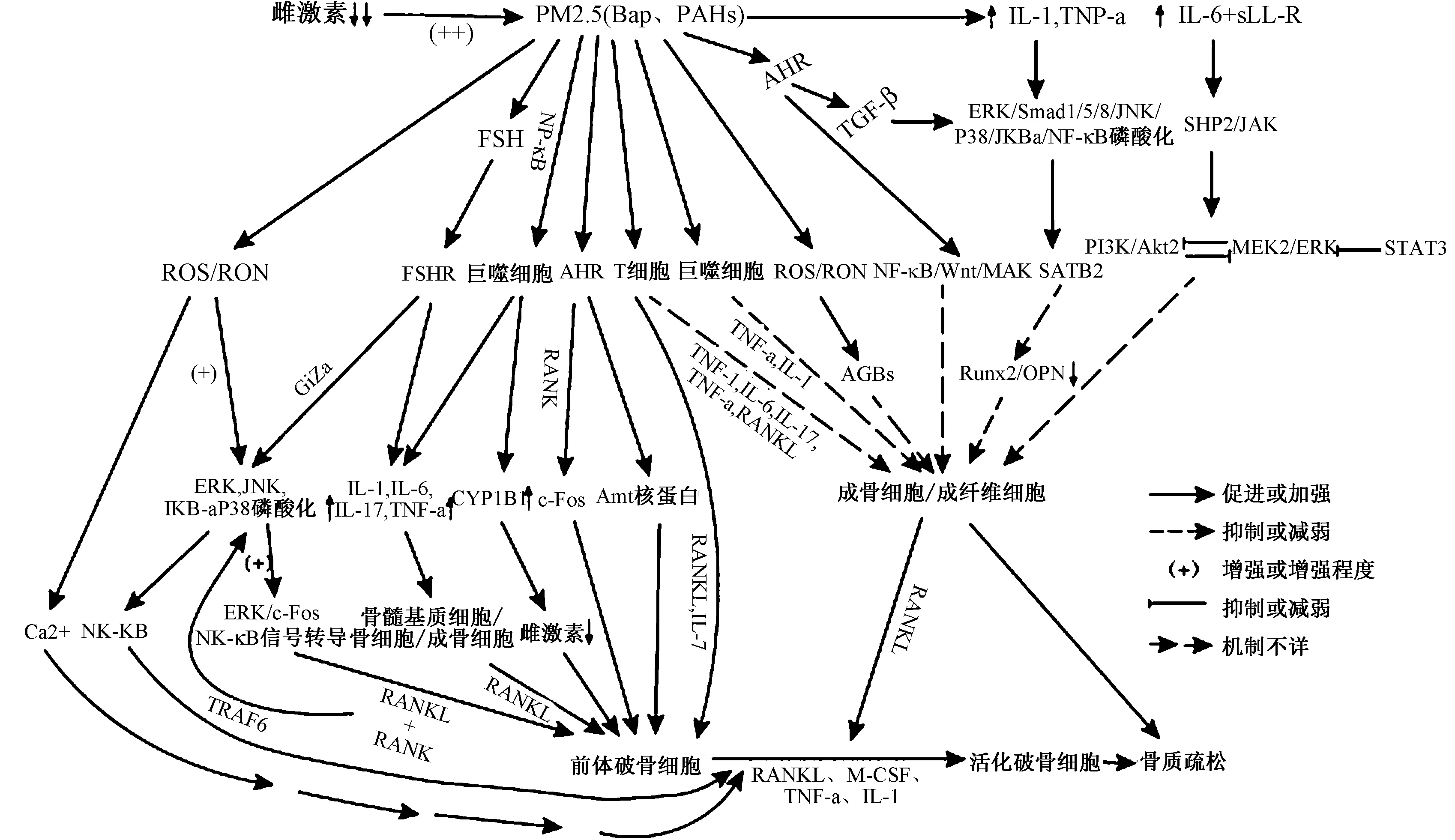
图1 PM2.5在低雌激素水平中对骨代谢影响的机制(具体机制详见文中参考文献)。Fig.1 The mechanism of PM2.5 affecting bone metabolism at low estrogen levels
1.3芳香烃受体(AhR)
芳香烃受体(AhR)广泛分布于人体组织器官中(如骨),与ER以复杂的方式相互起干扰作用,且具有类雌激素或抗雌激素样作用。TCDD、PAHs、BaP等是典型芳基烃受体(AhR)激动剂和CYP1A1诱导剂,并通过AHR与ERα之间相互干扰来共同竞争激活Arnt核蛋白,干扰雌激素信号传导,促进破骨细胞基因表达[44]。多环芳烃(PAHs)介导 NF-kB诱导巨噬细胞和破骨细胞表达CYP1B1(一种雌激素代谢酶)增加,使骨中雌激素水平降低,导致骨成积率降低,影响骨重塑[45]。Iqbal等[46]指出,破骨细胞作用在Cyp1a1/1a2/1b1-/-三重KO小鼠骨吸收减少,并且在AhR-/-的小鼠,骨量显著增加。Hžd′alová等[47]利用雌激素敏感的人乳腺癌MCF-7细胞的AhR基因敲除变异体,观察到芳香烃化合物及代谢物在MCF-7 AhRKO细胞中的雌激素样作用减弱,而CYP1A1和CYP1B1酶的异位表达部分恢复了BAP代谢及其对细胞增殖的影响。另外,在对敲除AhR(RANKΔOc/ΔOc)的骨髓巨噬细胞培养中,也发现B淋巴细胞诱导CYP1B1、CYP1A2表达下调,呈现破骨功能受损和分化抑制[48]。
此外,芳香烃化合物通过激活AhR来抑制SMAD依赖的信号通路TGF-β1/Smad4和SMAD非依赖的TGF-β1/ERK/AKT信号通路,使其下游相应蛋白磷酸化降低,成骨细胞基因Runx2和OPN明显降低,导致成骨分化和成熟均受抑制,骨生成减少[49]。同时,激活AhR还能通过RANK/c-Fos信号轴调控髓系前体向破骨细胞的分化,在AhR过表达的BMMS中,骨吸收面积和CTX-1的释放均明显升高,促进破骨作用[50]。激活AhR还可介导影响NF-κB、Wnt、MAPK等信号通路蛋白的转录促进破骨作用[51]。
2 其他介导途径
垂体-骨轴具有内分泌骨重塑调节的功能。垂体激素(FSH)能通过破骨细胞及其前体物上的Gi 2α偶联FSH受体(FSHR)来增强ERK、Akt、 IκBα(核因子κB-轻链增强子)的磷酸化,从而增强ERK/c-Fos和NF-κB信号转导,介导RANKL的促骨吸收作用[52]。另外,FSH可直接刺激骨髓微环境产生TNFα、IL-1β、IL-6,而FSHβ基因缺陷小鼠骨髓巨噬细胞产生炎症因子减少,即使雌激素缺乏,仍可免于骨量丢失[53-54]。PM2.5暴露诱导下丘脑炎症因子释放,从而抑制下丘脑-垂体-性腺轴,影响垂体LH和FSH释放,影响相应功能[55]。应用FSH特异性抗体可增加骨量[56]。
3 结语
PM2.5可通过多途径、多通路促进雌激素缺乏症的骨质丢失。近年来,国内有关学者越来越注意到环境污染对骨侵蚀的作用。笔者有望通过在体水平或细胞水平建模探讨PM2.5暴露对年龄相关性雌激素缺乏介导骨质丢失发生、发展的研究。
猜你喜欢
现代农业科技(2022年5期)2022-12-14
中国老年学杂志(2022年19期)2022-11-21
科学导报(2022年11期)2022-03-03
中国骨质疏松杂志(2022年1期)2022-02-17
中华骨与关节外科杂志(2021年12期)2021-08-31
天津医科大学学报(2021年3期)2021-07-21
天津医科大学学报(2021年3期)2021-07-21
山东医药(2020年36期)2020-12-31
智慧健康(2020年9期)2020-12-03
中国临床医学(2019年3期)2019-01-04
