
加替沙星的拉曼、红外光谱和密度泛函理论研究
2020-05-29 08:21:42辛敏思刘春宇蔡红星
光谱学与光谱分析 2020年5期
徐 笛,范 雅,辛敏思,刘春宇,张 烨,蔡红星
长春理工大学理学院,吉林 长春 130022
引 言
加替沙星(Gatifloxacin, Gati)是由日本杏林制药株式会社首创的新6-氟-8-甲氧基喹诺酮类外消旋化合物,是第四代氟喹诺酮类药物。主要用于由敏感病原体所致的轻、中度感染性疾病,因其广谱、高效、低毒的优越性,受到国内外广泛关注并使用。
加替沙星的分子量为375.39,分子式C19H22FN3O4,危险标识码Xn,属于有害物质。除了临床使用有诸多禁忌外,使用后还可能存在药物残留,危及健康。所以快速检测鉴定便尤为重要。目前常用的主要有高效液相色谱法(HPLC),荧光光度法等。但HPLC法的实验成本高,样品前处理较为复杂,液相色谱仪价格及日常维护费用比较昂贵,不利于实现快速检测。
拉曼光谱与红外光谱法具有无损快速、检出限低、且光谱信息互补的优越性,越来越多的在食品、药品检测中得到应用。密度泛函理论(density functional theory,DFT)是计算振动光谱的一种理论方法,其优势在于不明显增加计算量的同时,又考虑到了电子相关[1-2]。因其能够直观反应分子振动信息,是量子化学计算常用的方法[3-4]。目前以密度泛函计算为基础、与拉曼光谱相结合来分析物质结构信息的研究工作在文献中多有报道[5-6]。但还没有关于加替沙星的IR,Raman和DFT的比较研究。
1 实验部分与理论计算
加替沙星选用阿拉丁试剂官网的分析纯药品(按C19H22FN3O4计,含量>98%); LabRam HR Evolution型拉曼光谱仪(HORIBA公司),选择532 nm激光为激发光源,激光输出功率为31.675 mW,扫描时间10 s,探测器采用研究级大芯片尺寸空冷CCD; Thermo Scientific Nicolet iS50型傅里叶红外光谱仪,光谱分辨率4 cm-1,加替沙星粉末由KBr压片处理,扫描32次。
加替沙星的理论计算采用Gaussian09[7]软件包,分子构型由Gaussian view 5.0构建。由于加替沙星分子主要由C,H,O和N等轻元素构成,而Beckes三参数混合模(B3LYP)泛函在轻元素构成的分子计算中被广泛应用[5-6],故利用B3LYP泛函来算加替沙星的拉曼光谱与红外光谱。
首先利用B3LYP/3-21G基组对初始结构进行粗优化,在得到的优化结构的基础上,选择B3LYP/6-311+G(d)基组进行再优化并计算拉曼与红外光谱。理论光谱的频率修正选择6-311+G(d)基组的校正因子0.977[8],修正后再与实验数据相比较。计算结果无虚频,说明得到的是稳定结构。优化后得到的加替沙星分子结构和各个原子的名称与编号如图1所示。同时给出加替沙星分子优化后的空间几何参数,包含键长、键角和二面角,详见于表1。图2给出Gati分子的理论拉曼光谱(a)与红外光谱(b)的对比; 而理论拉曼光谱(c)与实验拉曼光谱(d)的对比如图3所示出,理论红外光谱(e)与实验红外光谱(f)的对比如图4所示。

图1 在B3LYP/6-311+G(d)基组优化后加替沙星结构Fig.1 Optimized structure of Gatifloxacinat B3LYP/6-311+G(d) levels
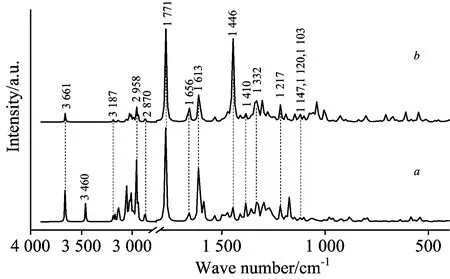
图2 400~4 000 cm-1范围内加替沙星的理论拉曼与红外光谱Fig.2 DFT-RS (a) and DFT-IR (b) of Gatifloxacin inthe 4 000~400 cm-1 region
2 结果与讨论
2.1 Gati分子的空间几何结构
通过Gaussian09优化后的Gati分子为三维非平面结构。如图1所示,Gati分子结构主要由以喹啉环为主体,1C上连接一个哌嗪环,2C与33N上分别连接一个甲氧基与环丙基,9C上连接一个羧基; 哌嗪环14C上连接一个甲基。从表1中1C—2C—3C—4C二面角为9.381 096 0°、7C—9C—10C—33N二面角为-7.809 60°等发现喹啉环不是平面几何结构;14C—21N—16C—13C二面角为-28.671 47°、13C—20N—12C—14C二面角为-33.357 11°等证明哌嗪环同样不是平面环结构。

表1 加替沙星优化后的几何参数Table 1 Optimized geometrical Parameters of Gatifloxacin
2.2 光谱分析
通过Gaussian view 5.0观察Gati分子理论拉曼与红外光谱各谱峰的振动形式,对其振动模归属进行指认,整理归纳于表2。表中前2列分别是实验测得与DFT计算所得的红外光谱各谱峰的振动波数,中间2列分别是实验测得与DFT计算所得的拉曼光谱各谱峰的振动波数,每个谱峰所属的振动模式均在第5列中给出。

表2 加替沙星理论与实验振动频率(cm-1)与归属Table 2 Theoretical and experimental vibrational frequencies (cm-1) and assignments of Gatifloxacin
续表2

549541ρ(C—H)C3H5 δ(qu1) ν(C—F)515513512ω(C—C)qu1br(pi)485469ρ(C—H) C3H5 δ(qu2)452456σ(C—N)pi399392ρ(6C—F 7C—O 29C—O)374ρ(2C—19O) σ(C—30O)339ρ(14C—23C)328312ω(C—19O)
Note: (1)ν: Stretch;σ: Scissoring;ρ: In-plane-rocking;ω: Out-of-plane-rocking;τ: Torsion-vibration;δ: Deformation-vibration; br: Ringbreath; (2) s: Symmetric; as: Antisymmetric; (3) pi: Pierazine ring; qu: Quinoline
2.2.1 理论计算所得拉曼与红外光谱的对比分析
和大多数有机分子一样,加替沙星分子也具有不完全的对称性,因而在红外与拉曼光谱中都有反映,故对其理论计算所得的红外光谱与拉曼光谱(图3)进行对比分析。
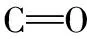

图3 加替沙星理论与实验拉曼光谱比较Fig.3 Comparison of DFT-RS (c) and NRS (d) of Gati

图4 加替沙星理论与实验红外光谱比较Fig.4 Comparison of DFT-IR (e) and IR (f) of Gati

2.2.2 理论光谱与实验光谱的对比分析
将理论计算所得的光谱与实验测得的光谱进行对比,如图4与图5所示。实验测得的拉曼光谱与红外光谱谱峰数量多、强度明显,说明实验结果较好。通过比较理论光谱与实验光谱的光谱线型和振动频率发现,大部分谱峰的峰位基本一致。如哌嗪环上16C—H的对称伸缩振动在实验红外光谱中位于3 012 cm-1,理论红外光谱中位于3 011 cm-1; 实验拉曼光谱中位于3 016 cm-1,理论拉曼光谱中位于3 011 cm-1。喹啉环上C—C伸缩振动在实验光谱中位于1 616 cm-1(IR),1 614 cm-1(NRS),理论红外和拉曼光谱中都位于1 613 cm-1。
个别谱峰存在差异,这些差异主要体现在两方面: 其一为相应峰位不一致: 如环丙基上C—H的面内摇摆振动在实验光谱中都位于1 093 cm-1,而在理论光谱中都位于1 103 cm-1,峰位波数相差6 cm-1。其原因可能是实验设备会产生随机误差,同时量子化学的计算中过多考虑了电子相关的影响。其二是理论光谱中存在的个别谱峰在实验光谱中没有测到: 如理论光谱中1 771 cm-1等处的谱峰在实验红外和拉曼光谱中都没有观测到。这可能是由于理论计算模拟纯理论振动,而实验中加替沙星以固体粉末形式存在,有分子间作用力影响。
3 结 论
拉曼光谱是由具有对称分布的键的对称振动引起,而红外光谱是由分子的不对称振动所引起。利用这两种光谱信息互补的特性,能够实现有机化合物种类和结构的准确判断。采用密度泛函理论的方法,结合Gaussian可视化软件,对加替沙星的分子结构进行了优化,计算出其拉曼光谱与红外光谱,确定了各谱峰的振动模式归属,并与加替沙星分析纯药品的实验光谱进行了对比分析。该研究为新型喹诺酮类抗生素的振动光谱检测储备了基础数据,为其在药品残留检测领域的应用提供参考。
猜你喜欢
探测与控制学报(2023年4期)2023-09-12 07:26:12
分析科学学报(2021年3期)2021-07-14 01:51:16
色谱(2021年6期)2021-05-06 02:18:56
科技资讯(2020年12期)2020-06-03 04:44:20
大东方(2017年10期)2017-05-30 18:29:24
国外医药(抗生素分册)(2016年5期)2016-07-12 14:25:34
国外医药(抗生素分册)(2016年2期)2016-07-12 14:25:01
兽医导刊(2016年12期)2016-05-17 03:51:52
郑州大学学报(理学版)(2013年3期)2013-03-11 20:30:38
化学分析计量(2013年1期)2013-03-11 16:37:15
