
啶虫脒固体制剂中违禁添加氟虫腈的红外光谱定量方法研究
2020-05-19 15:23杜夏瑜熊艳梅夏静静闵顺耕
分析化学 2020年4期
杜夏瑜 熊艳梅 夏静静 闵顺耕
摘 要 利用红外光谱结合衰减全反射技术(Attenuated total reflection Fourier transform infrared spectroscopy,ATR-FT-IR)对啶虫脒固体制剂中氟虫腈的含量进行快速测定。采用傅里叶变换红外光谱仪采集69个固体样品及其萃取液的光谱,探究不同预处理方法及变量选择方法对样品光谱的处理效果,建立固体样品及萃取液的定量分析模型。通过Kennard-Stone算法划分样本集,采用偏最小二乘法建立氟虫腈的定量模型,使用外部检验对模型进行评价。结果表明,对于固体样品,直接测定其中氟虫腈的定量模型的决定系数为0.9762,预测均方根误差(Root mean square error of prediction set,RMSEP)为0.0022;经N,N-二甲基甲酰胺(DMF)萃取,上清液样品中氟虫腈定量模型的预测决定系数为0.9810,RMSEP为0.0019;对于氟虫腈含量范围在060%~5.00%的样品,外部检验样品预测平均相对误差(MRE)为0.08。本方法可应用于农药中违禁添加氟虫腈的現场快速检测。
关键词 红外光谱;偏最小二乘法;变量选择;氟虫腈;啶虫脒
1 引 言
氟虫腈属于GABA-氯离子通道抑制剂,与常用的多类杀虫剂无交互抗性,杀虫谱广,但对蜜蜂毒性高,且在水体和土壤中降解缓慢,环境友好性差[1,2]。啶虫脒是一种新烟碱类杀虫剂,具有较强的杀虫活性,作用于昆虫神经系统,可有效防治大部分产生抗性的害虫。2009年2月25日农业部发布公告,对氟虫腈的使用进行了限制,规定含氟虫腈成分的农药制剂仅能申请卫生用、玉米等部分旱田种子包衣剂的田间试验、农药登记和销售使用[3]。但由于氟虫腈在啶虫脒制剂中混合度较好,二者使用条件相似,一些不良商家和企业将氟虫腈非法添加到啶虫脒制剂中以增强施用效果,这些含有氟虫腈的农药制剂的使用对人畜、环境等带来许多不良影响,迫切需要开发一种快速检测氟虫腈含量的方法。
常规的氟虫腈检测方法为色谱法或色谱-质谱联用法[4~8],这类方法需要的时间较长,且对样品的前处理要求较高,操作繁琐,难以适用于现场快速检测。红外光谱技术具有测定速度快、检测成本低、对前处理要求低、非破坏性检测等特点,在食品、农业、化学、医药、纺织等行业应用广泛[9],结合化学计量学在数据处理方面具有很大优势,近几年应用于农药有效成分及违禁成分的检测[10,11]。Li 等[12]利用红外光谱结合区间组合优化(Interval combination optimization,ICO)算法建立了啶虫脒乳油中氟虫腈含量的定量模型,最优模型的决定系数为0.9982;但未涉及啶虫脒固体制剂中氟虫腈含量的定量测定,且乳油的基质相对简单。
目前,对氟虫腈红外光谱定量分析的算法一般为偏最小二乘法(Partial least squares,PLS),其建模方法对校正集要求较高,建模时间长[12],因此,在建模前需要划分样本,并对原始光谱进行预处理和变量选择,采用Kennard-Stone (KS)法划分样本,以保证校正集样本按照空间距离分布均匀[13]。 预处理方法包括平滑去噪算法(Smoothing)、Savitzky-Golay导数法(Savitzky-Golay derivative)、标准正态变换法(Standard normal variation,SNV)、多元散射校正法(Multivariate scattering correction,MSC)等[14~16],变量选择方法包括区间偏最小二乘法(Interval partial least-squares,iPLS)、移动窗口偏最小二乘法(Moving window partial least squares,MWPLS)、ICO等[17~19]。
本研究采用红外光谱结合衰减全反射技术(ATR-FT-IR)对啶虫脒固体商品制剂中违禁添加的氟虫腈含量进行检测,分别采用固体直接测定和N,N-二甲基甲酰胺(DMF)溶剂萃取液测定方法,对样品中的氟虫腈进行定量检测;采用4种光谱预处理方法、3种变量选择方法对模型进行优化,采用PLS法建立最优定量模型。所建立分析方法可应用于农药违禁添加氟虫腈的现场快速检测。
2 实验部分
2.1 仪器与试剂
Nicolet iS5TM型FT-IR光谱仪(美国Thermo Fisher公司,配有Omni iD5-ATR金刚石附件,仪器参数为:扫描范围4000~650 cm 1,分辨率2 cm 1,扫描次数32次);SCDEALL VX-Ⅲ型多管涡旋振荡器(安简(北京)科技有限公司);TG16MW型台式高速离心机(湖南赫西仪器装备有限公司)。
40%啶虫脒可湿性粉剂(青岛瀚生生物科技股份有限公司;农药登记证号:PD20096672);70%啶虫脒可湿性粉剂(江西禾益化工股份有限公司;农药登记证号:PD20142282);40%啶虫脒可溶粉剂(海南正业中农高科股份有限公司;农药登记证号:PD20102099);氟虫腈原药(95%,w/w,中国农科院植保所农药厂);乙腈、无水乙醇、DMF(分析纯,北京化工厂);所有试剂均未经过纯化,直接使用。所有的玻璃器皿及仪器可清洗部分均分别使用去离子水和无水乙醇清洗3遍,干燥后使用。
2.2 数据处理软件
10.3版The Umscrambler X、9.2版OMNIC、R2019a 版Matlab、2019版Excel和9.1版Origin。
2.3 光谱采集及数据处理
2.3.1 样品制备 在3种不同来源的啶虫脒制剂中加入不同质量的95%(w/w)氟虫腈原药,研细后,置于涡旋振荡器上振荡5 min,配制氟虫腈浓度在0.1%~5.0%(w/w)之间的固体样品,共69个,样品质量百分数均值为1.315%,用于建立定量模型和验证。
分别用乙腈、甲苯、二甲苯和DMF 4种溶剂对适量氟虫腈原药进行溶解,其中DMF对氟虫腈的溶解效果最好,故选用其作为固体样品的萃取溶剂,对上述69个样品分别萃取:分别称量适量样品,加入1 mL萃取液,置于涡旋振荡器上振荡5 min,4000 r/min离心3 min,得到69个上清液,样品质量百分数均值为1.344%,用于建立定量模型和验证。
2.3.2 光谱采集 使用FT-IR光谱仪采集69个样品及对应的69个上清液的红外光谱。每个样品采集3条光谱并求平均值,以氟虫腈在啶虫脒制剂中的含量作为分析指标进行处理。
2.3.3 数据处理 对样品和上清液的光谱数据分别建立定量模型,其步骤概述为:基线校正预处理光谱后,采用4种预处理方法(平滑去噪、Savitzky-Golay导数、SNV、MSC)结合3种变量选择方法(iPLS、MWPLS、ICO)优化定量模型。
实验中用到的基线校正在The Umscrambler X中进行,其余所有光谱处理的算法代码均在Matlab中完成。模型评价采用RMSEP为指标。
3 结果与讨论
3.1 氟虫腈、啶虫脒制剂及样品的衰减全反射中红外光谱图
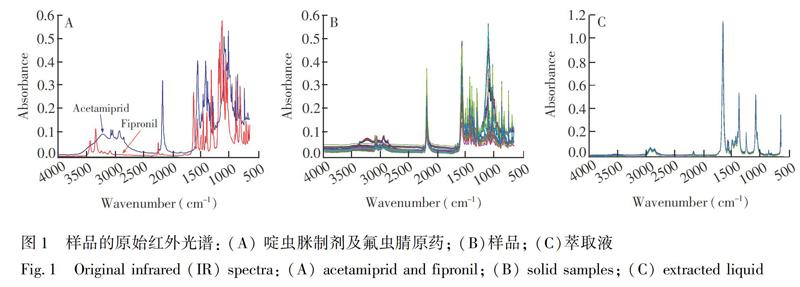
图1为氟虫腈、啶虫脒制剂及样品的红外谱图。氟虫腈原药在3360~3320 cm 1处有中强度的吸收峰,为NH伸缩振动;2240 cm 1处的吸收峰为CN伸缩振动;1690~1630 cm 1处的强吸收峰为CN的伸缩振动;1590 cm 1处中强度的吸收峰为NH的变形振动;1360~1320 cm 1处中强度的吸收峰为NCN的反对称伸缩振动;1190~1140 cm 1处中强度的吸收峰为NCN的对称伸缩振动;865~810 cm 1处的吸收峰为四取代苯环的特征吸收峰。啶虫脒制剂在3200 cm 1处的宽峰为制剂中OH的吸收;2250~2100 cm 1处的强吸收峰为硅藻土基质SiH伸缩振动;1580 cm 1处的中强吸收峰应为制剂基质中的胺类或双键物质;1360~1310 cm 1处的强吸收峰为CN的伸缩振动。
1处的峰被啶虫脒制剂的基质吸收掩盖,因此对样品进行萃取以排除基质的干扰。由图1可见,萃取液的红外光谱背景更干净,谱图质量有所改善。
3.2 固体样品中氟虫腈定量模型的建立及优化
对69个固体样品对应的69条光谱,以啶虫脒制剂中氟虫腈含量为化学值,通过KS法划分校正集、验证集样品分别为46和23个,采用PLS法建立氟虫腈含量的定量模型。
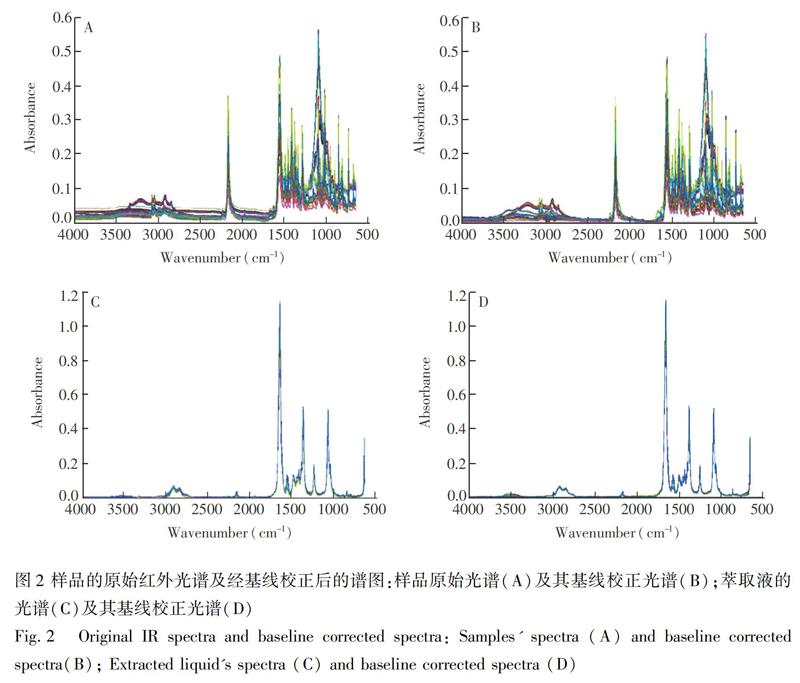
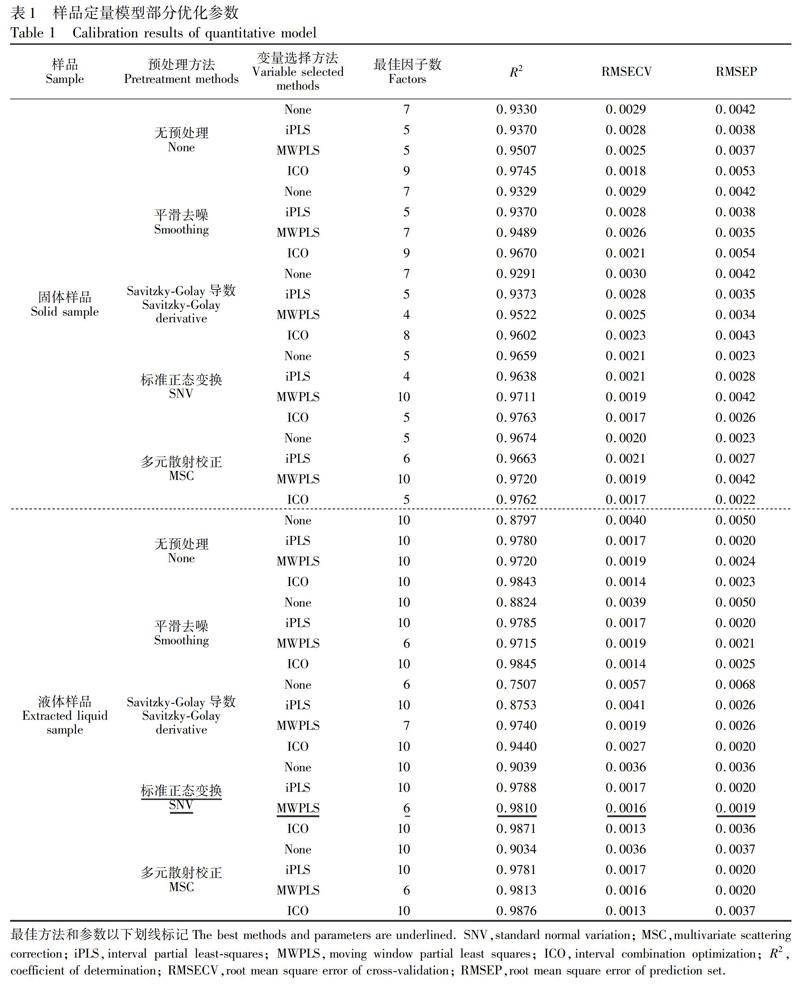
进行模型优化,对光谱进行基线校正处理,得到光谱图见图2。由图2可见,基线校正显著消除了基线处的部分噪声干扰。采用4种预处理方法(平滑去噪、Savitzky-Golay导数、SNV、MSC)结合3种变量选择算法(iPLS、MWPLS、ICO)优化模型,得到最优模型为MSC预处理结合ICO方法选择的变量建立的模型,其参数为:R2=0.9762,RMSECV=0.0017,RMSEP=0.0022(模型参数见表1)。ICO选择的变量见图3,选择了波数在(a)3920~3830 cm 1、(b)3750~3580 cm 1、(c)2238~2165 cm 1、(d)1389~1310 cm 1、(e)1140~1070 cm 1和(f)908~813 cm 1的区域,其中: (a)和(b)两区域无明显吸收;(c)区域在2170 cm 1处有一个明显吸收峰,是氟虫腈的CN结构信息,峰高随氟虫腈含量变化明显;(d)和(e)两区域的多组峰包含了氟虫腈NCN结构的振动信息,且峰高随氟虫腈含量变化明显;(f)区域包含氟虫腈四取代苯环骨架的特征吸收,其峰高随氟虫腈含量变化明显,以上4个区域选择的光谱范围见图4。模型的预测值与化学值相关关系图见图5,优化后的模型对样品的预测效果更好。
3.3 固体样品中氟虫腈定量模型的验证
将KS采样法选择出的23个样品作为验证集,预测结果见表2。由表2可知,对于含量在0.10%~5.00%的样品,预测结果的相对误差均低于0.58;具体而言,对含量在0.10%~0.60%的样品预测误差较大,预测平均相对误差(MRE)为0.37;对含量在0.60%~5.00%的样品预测效果较好,MRE为0.10。
3.4 萃取液中氟虫腈最优定量模型的建立及验证
为进一步优化3.2节建立的定量模型,考虑到啶虫脒制剂固体基质较为复杂,且不同固体制剂的基质各不相同,对有效成分低的样品测定干扰较大,适用范围有限。因此,用溶剂DMF萃取固体样品中的氟虫腈并采集光谱,以氟虫腈在啶虫脒制剂中的含量为分析指标建立模型:对光谱进行基线校正(见图2),显著消除了液体样品在基线处的噪声;采用4种预处理方法(平滑去噪,Savitzky-Golay导数,SNV,MSC)结合3种变量选择算法(iPLS,MWPLS,ICO)优化模型。
得到最优模型为SNV预处理结合MWPLS方法选择的变量建立的模型,其参数为:R2=0.9810,RMSECV=0.0016,RMSEP=0.0019(模型參数见表1)。MWPLS选择的变量见图3,选择了波数在(g). 1335~1300 cm 1范围的区域,该区域光谱见图4。由图4可见,区域内1315 cm 1处有一个吸收峰,峰高随氟虫腈含量变化明显,代表了氟虫腈N-C-N的振动。模型的预测值与化学值的相关关系图见图5,可见萃取液建立的模型对样品的预测效果最优。
将KS采样法选择出的23个样品作为验证集,预测结果见表2。对于含量在0.10%~5.00%的样品,预测结果的相对误差均低于0.78;具体而言,对含量在0.10%~0.60%的样品预测误差较大,MRE为0.49;对含量在0.60%~5.00%的样品预测效果较好,MRE为0.08。与固体样品模型的预测能力相比,对于低含量(0.10%~0.60%)样品的预测能力较差,可能是由于低含量样品含量过低,称量及萃取过程中存在较大误差,且红外吸收受萃取溶液溶剂峰的影响过大;对于较高含量(0.60%~5.00%)样品的预测能力有较大提高,预测更稳定。
4 结 论
本研究测定了固体农药制剂中氟虫腈的含量及其经萃取后液体样品中的氟虫腈含量,得到的模型对氟虫腈含量在0.60%~5.00%的啶虫脒商品制剂含量预测效果较好,MRE为0.08,在《农药制剂产品中微量其他农药成分限量》团体标准[20]允许的含量(<5%)范围内,能满足实际农药质量监管中氟虫腈的分析要求,为固体农药混合制剂中有效成分的检出提供了理论基础,但对于低含量样品(0.10%~0.60%)有效成分的快速准确检测仍需进一步的探索。
References
1 Tingle C C D,Rother J A,Dewhurst C F. Rev. Environ. Contam. Tokicol.,2003,176: 1-66
2 EI Hassani A K,Dacher M,Gauthier M,Armengaud C. Pharmacol. Biochem. Behav.,2005,22(1): 68-102
3 http://www.moa.gov.cn/govpublic/ZZYGLS/201006/t20100606_1534288.htm
农业部第1157号公告No. 1157 Announcement of Ministry of Agriculture
4 Hafeez A,Tawab I A,Iqbal S. J. AOAC Int.,2016,99(5):1185-1190
5 Zhang M,Bian K,Zhou T,Song X,Liu Q,Meng C. J. Chromatogr. B,2016,1014: 31-36
6 Jimenez J J,Bernal J L,del Nozal M J,Martin M T,Mayo R. J. Chromatogr. A,2007,1146(1): 8-16
7 NING Xiao,JIN Shao-Ming,GAO Wen-Chao,CAO Jin,DING Hong. Chinese J. Anal. Chem.,2018,46(8): 1297-1305
宁 霄,金绍明,高文超,曹 进,丁 宏. 分析化学,2018,46(8): 1297-1305
8 ZHU Li-Ping,ZHU Tao,PAN Yu-Xiang,SUN Jun,DONG Jing. Chinese J. Anal. Chem.,2008,36(7): 999-1003
朱莉萍,朱 涛,潘玉香,孙 军,董 静. 分析化学,2008,36(7): 999-1003
9 Antony B,Mehta B M,Sharma S,Ratnam K,Aparnathi K D. J. Food Sci. Tech.,2018,55(9): 3632-3639
10 WU Rui-Mei,WANG Xiao,GUO Ping,AI Shi-Rong,YAN Lin-Yuan,LIU Mu-Hua. Journal of Instrumental Analysis,2013,32(11): 1359-1363
吴瑞梅,王 晓,郭 平,艾施荣,严霖元,刘木华. 分析测试学报,2013,32(11): 1359-1363
11 WU Hou-Bin,YU Rong,LIU Ping-Ping,MU Lan,LIU Feng-Mao,ZHANG Yan-Qiu,JIANG Shu-Ren,MIN Shun-Geng. Agrochemicals,2012,51(4): 270-272
吴厚斌,于 荣,刘苹苹,穆 兰,刘丰茂,张延秋,江树人,闵顺耕. 农药,2012,51(4): 270-272
12 Li Q,Huang Y,Song X,Zhang J,Min S. Pest. Manag. Sci.,2019,75(6): 1743-1749
13 Kennard R W,Stone L A. Technometrics,2012,11(1): 137-148
14 Peter C,Grace W. Numer. Math.,1978,31(4): 377-403
15 Ruffin C,King R L,Younan N H. Gisci. Remote Sens.,2008,45(1): 1-15
16 Dhanoa M S,Lister S J,Sanderson R,Barnes R J. J. Near Infrared Spectrosc.,1994,2(1): 43-47.
17 Norgaard L,Saudland A,Wagner J,Nielsen J P,Engelsen S B. Appl. Spectrosc.,2000,54(3): 413-419
18 Jiang J H,Berry R J,Siesler H W,Ozaki Y. Anal. Chem.,2002,74(14): 3555-3565
19 Song X,Huang Y,Yan H,Xiong Y,Min S. Anal. Chim. Acta,2016,948: 19-29
20 T/CCPIA 022-2019,Limit of Trace Amounts of Other Pesticide Ingredients in Pesticide Formulation Products. Association Standards of the People's Republic of China
農药制剂产品中微量其他农药成分限量. 中华人民共和国团体标准. T/CCPIA 022-2019
