
雪菊中木犀草苷的富集及近红外光谱分析方法
2020-05-19 15:23楚刚辉王坤尹学博
分析化学 2020年4期
楚刚辉 王坤 尹学博


摘 要 采用温和的方法制备了苯硼酸吸附剂,将其用于雪菊中微量木犀草苷的富集,并以此为基础建立了快速、选择性地测定雪菊中木犀草苷含量的近红外光谱分析方法。富集了木犀草苷的吸附剂无需脱附,即可用近红外漫反射光谱直接检测,通过偏最小二乘回归法建立了定量分析木犀草苷的校正模型,并成功实现了木犀草苷含量的测定。采用此苯硼酸吸附剂0.2 g,常温振荡吸附20 min后,吸附效率可达85.5%。结果表明,经连续小波变换处理近红外光谱分析数据,木犀草苷预测模型的预测浓度和参考浓度之间的相关系数为0.9765,且木犀草苷在0.15~19.5 mg/L浓度范围内具有较好的预测结果,预测回收率为84.7%~113.2%。本研究建立了苯硼酸吸附剂吸附预富集方法,与近红外漫反射光谱结合,实现了雪菊溶液中微量木犀草苷的选择性测定,为近红外光谱与吸附富集分析药食用植物中的其它有效成分的研究提供了参考。
关键词 木犀草苷;苯硼酸吸附剂;富集;选择性检测;近红外漫反射光谱
1 引 言
木犀草苷(Luteoloside)是一种天然植物黄酮类物质,为弱酸性四羟基黄酮类化合物。木犀草苷又名木犀草素-7-O-葡萄糖苷(Luteolin-7-O-glucoside),其苷元为木犀草素,因最初是从木犀草科木犀草属植物木犀草中分离出而得名。木犀草苷具有抗炎、抗病毒、抗肿瘤和降低胆固醇等药理作用[1,2]。雪菊是一种双色坚果菊花,作为一种药食两用植物,药理研究证实雪菊具有极高的药用价值。雪菊的有效成分包括黄酮类、糖类、矿物质元素、酚类等,具有清热解毒、活血、养胃、健脾、抗疲劳和抗氧化等功效[3~5]。植物样品和中药中木犀草苷的常见检测方法包括高效液相色谱法[6,7]、超高效液相色谱-质谱联用法[8]、高效液相色谱-质谱联用法[9]和超临界色谱法[10]等。这些方法选择性好,但不同程度上存在有机试剂消耗量大、操作过程繁琐、分析时间长等问题。
近红外光是介于中红外和可见光之间的电磁波,其波长范围为780~2526 nm[11],近红外光谱技术可根据有机物含氢基团(主要包括CH、NH、OH、SH等)对近红外光谱吸收的差异,区分不同物质并测定其浓度[12],具有无损、简单、方便、处理量大、快速、准确、环境友好等优点[13]。自20世纪90年代以来,近红外光谱技术逐漸应用在生产和基础研究中 [14,15]。药食两用植物属于复杂体系,选择性检测其中的有效成分一直是相关研究的热点,而近红外光谱可用于复杂体系成分分析[16,17]。由于近红外光谱的检出限高,待测组分的含量一般应大于0.1%。为提高近红外光谱的检出限和分析灵敏度,文献报道了多种吸附材料,用于样品中微量成分的富集及近红外光谱检测[18~20]。为了提高检测的灵敏度和选择性,本研究基于含苯硼酸基团的化合物与多元醇化合物的共价化学键特异性结合的性质[21,22],采用温和的方法制备了苯硼酸吸附剂,用于富集雪菊中微量的木犀草苷,采用近红外光谱结合化学计量学方法实现了其中木犀草苷的选择性检测。
2 实验部分
2.1 仪器与试剂
SF-TDL-2SOA低速离心机(上海菲恰尔分析仪器有限公司);SJZ-82电热恒温水浴锅(天津市泰斯特仪器有限公司);DZF-6050真空干燥箱(上海齐欣科学仪器有限公司);HY-2型多用调速振荡器(江苏省金坛医疗仪器厂);AntarisⅡ近红外光谱仪(美国Thermo公司);TU-1900 型双光束紫外可见分光光度计(北京普析通用仪器有限公司);DF-4型压片机(天津市港东科技发展有限公司);JJ-2增力电动搅拌器(金坛市医疗仪器厂)。
3-氨基苯硼酸(3-Aminophenylboronic acid,APB)、3-缩水甘油基氧基丙基三甲氧基硅烷(3-Glycidyloxypropyltrimethoxysilane,GLMYO)、硅酸四乙酯(Tetraethyl orthosilicate,TEOS)、木犀草苷(上海阿拉丁生化科技股份有限公司);甲醇、氨水和无水乙醇均为分析纯试剂。选择产于新疆巴楚的雪菊,经粉碎机粉碎后备用。
2.2 实验方法
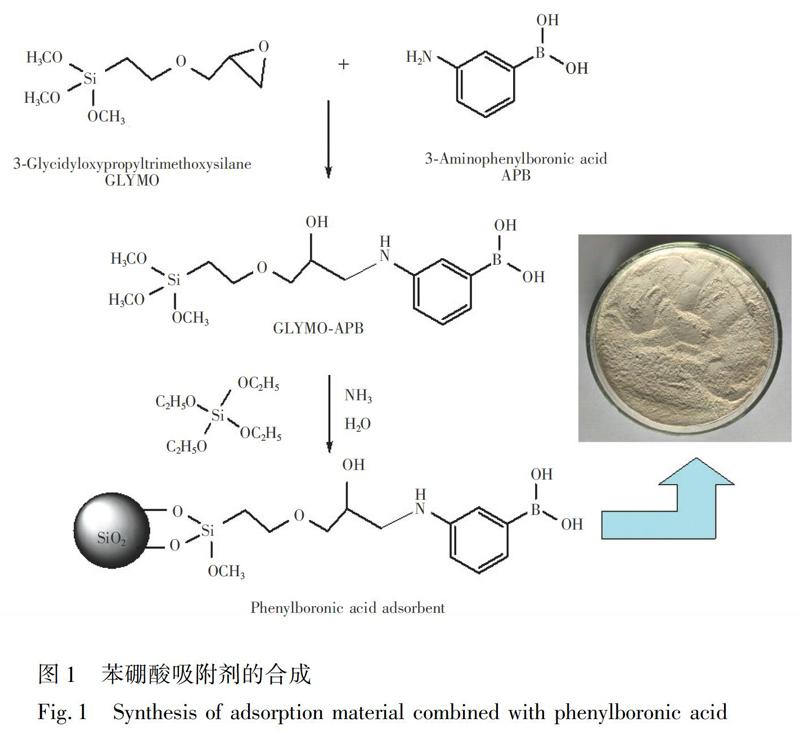
2.2.1 苯硼酸吸附剂的制备 参考文献[23]的方法,并做适当修改。取1.16 g APB、80 mL甲醇及2.0 mL GLYMO于三颈圆底烧瓶中,在40℃恒温水浴中电动搅拌反应12 h,得到GLYMO-APB。然后加入6.0 mL TEOS、2.0 mL纯净水、2.0 mL氨水,继续电动搅拌24 h(图1)。GLYMO-APB和TEOS在氨水条件下同步水解完成合成过程。反应完毕,将产物离心,先用水洗至中性,再用甲醇洗涤一次,收集固体样品,于真空干燥箱中60℃烘干24 h,干燥后研细,备用。
2.2.2 木犀草苷的吸附实验 以木犀草苷为研究对象,称取0.20 g苯硼酸吸附剂于100 mL锥形瓶中,加入50.0 mL木犀草苷/雪菊提取溶液,在室温下,于振荡器中振荡20 min,经量筒式过滤器过滤,测定滤液在349 nm的吸光度,木犀草苷的吸附率(R,%)的计算公式如下:
式中,A0和A分别代表木犀草苷溶液吸附前后的吸光度。
2.2.3 木犀草苷的HPLC测定法 采用HPLC法进行供试雪菊样品中木犀草苷的含量测定。 色谱条件: Agilent HC-C18色谱柱(250 mm ×4.6 mm,5 μm),流动相A为乙腈,B为0.2%醋酸溶液,梯度洗脱: 0~40 min,15%~90% A;40~50 min,90%A。流速: 1 mL/min;柱温,常温;进样量: 10 μL;检测波长: 349 nm。配制0.5~590 mg/L木犀草苷系列标准溶液,分别取10 μL,按上述色谱条件分析,获得木犀草苷色谱峰的峰面积。以峰面积为纵坐标,质量浓度(mg/L)为横坐标,得线性回归方程: y=76330x-2×106(R2=0.9820)。 雪菊样品溶液采用微孔滤膜(0.45 μm)过滤,取续滤液,进行色谱分析,木犀草苷标准样和雪菊提取液的色谱图见图2。
2.2.4 雪菊分析试液的制备与木犀草苷的富集 分别称取适量的巴楚雪菊于100 mL锥形瓶中,加入50 mL 35% (V/V)甲醇浸泡过夜(12 h);抽滤,收集上清液于干净锥形瓶中,备用。按照雪菊样品色谱检测的浓度结果,制成系列不同木犀草苷浓度的雪菊提取液68组,浓度范围为1.5~19.5 mg/L。称取0.2 g苯硼酸吸附剂于100 mL一系列锥形瓶内,将制备的68组雪菊提取溶液每组50 mL依次加入其中。于常温下振荡20 min,实现木犀草苷的富集,抽滤得68个富集有木犀草苷的吸附剂固体。富集有木犀草苷的吸附剂无需脱附,直接用于近红外光谱检测,得到68个样本的近红外光谱,其中39个用于建立定量校正模型,29个(其中12对重复样本)作为预测模型建立近红外光谱分析方法。
2.2.5 光谱测量方法 68个固体样本通过近红外积分球漫反射测量得到近红外光谱,选择仪器分辨率为4 cm1,光谱扫描范围为4000~10000 cm1。于室温下开机预热1 h后进行测量,测量时,每隔1 h,背景校正1次。为了减小误差,68个样本进行随机测量;为提高样品测定的信噪比,每个样品近红外光谱均经过64次扫描,并重复3次测量,以3次结果的平均值作为最终光谱。本研究联合多变量校准法和偏最小二乘回归(Partial least square regression,PLSR)进行分析,并根据仪器软件包TQ analyst 9实现建模和预测。
3 结果与讨论
3.1 苯硼酸吸附剂的红外光谱
为验证苯硼酸的成功接枝,将二氧化硅和苯硼酸吸附剂与KBr按质量比1∶100~1∶200压片,红外光谱见图3。通过比较二氧化硅的红外光谱,苯硼酸吸附剂在1357 cm 1显示BO键的伸缩振动峰,1506和1598 cm 1处有苯环的特征吸收峰,2943和2880 cm 1处属于甲基和亚甲基的伸缩振动峰。与二氧化硅红外光谱比较,这些新出现的吸收峰表明苯硼酸已经结合在二氧化硅表面。
3.2 苯硼酸吸附剂对木犀草苷的吸附
为确定苯硼酸吸附剂对木犀草苷的吸附性能,以10 mg/L 木犀草苷标准溶液为目标溶液,按照2.2.2节的方法进行吸附实验。采用紫外-可见光谱检测吸附前后木犀草苷溶液的光谱,计算吸附率(图4),其中349 nm为木犀草苷的特征吸收波长。研究表明,苯硼酸可与含有顺式二醇结构的邻苯二酚类物质发生特异性结合作用[24,25]。比较10 mg/L木犀草苷标准溶液的吸收光谱(a曲线)和经苯硼酸吸附剂吸附后的吸收光谱(b曲线),349 nm处的吸收明显减少,计算得到木犀草苷吸附率为85.5%。結果表明,苯硼酸吸附剂对木犀草苷具有良好的吸附作用。
按照2.2.4和2.2.5节的方法对吸附了木犀草苷的吸附剂进行光谱检测,得到了68个固体样品的近红外光谱(图5)。根据仪器软件包TQ analyst 9,选择的波数范围为4115~8115 cm 1。
3.3 木犀草苷的近红外光谱定量结果
3.3.1 木犀草苷定量校正模型的建立 通过PLSR法建立木犀草苷定量模型,为了优化定量校正模型,选择不同的方法,如一阶导数(First derivative,1st)、标准正态变换(Standard normal variate,SNV)、多元散射校正(Multiplicative scatter correction,MSC)、连续小波变换(Continuous wavelet transform,CWT)、移动窗口最小二乘多项式平滑(Savitzky-golay smoothing,SG)等光谱预处理方法[26,27],通过蒙特卡洛交叉验证(Monte carlo cross-validation,MCCV)法确定模型的因子数,表1为不同光谱预处理方法得到的木犀草苷PLSR定量校正模型及交叉验证结果。这些模型的因子数在8~12之间,因为近红外光谱中既包括了木犀草苷的信息,也包含有苯硼酸吸附剂和雪菊中其它复杂化合物的组分信号。采用PLSR方法,在样品中提取木犀草苷的定量信息,以3个参数综合评价木犀草苷PLSR定量模型的预测能力,分别为相关系数(Related coefficient,R)、交叉验证均方根误差(Root mean square error of cross validation,RMSECV)和预测残差值(Residual predictive deviation,RPD)。R反映木犀草苷的预测浓度与真实浓度的相关程度,RMSECV反映预测浓度与实际浓度的偏离程度。前期文献结果表明,当模型的RPD>2.5时,该处理方法可用于定量预测,RPD越大,说明定量结果越准确[28,29]。
由表1可知,相比无处理方法,雪菊样品的近红外光谱经二阶导数(Second derivative,2nd)、MSC、SNV处理后,RPD值显示结果更差;而经1st、SG平滑、CWT、MSC+1st、SNV+1st处理后,RPD值更大,说明这些光谱预处理可有效改善定量结果。比较这5种方法与无处理方法中的R、RMSECV、RPD值,经CWT处理后的相关系数R值最大,且RMSECV值最小,RPD值最大,达到5.88。为了使光谱干扰更小、误差更小、预测更准确,达到最优的定量效果,本研究选用CWT方法对所有木犀草苷样品的近红外光谱进行处理。结果表明,通过预富集和CWT方法可以提取样品中待测组分木犀草苷的有效检测信息。
3.3.2 木犀草苷定量模型的验证 使用29个预测集样品(12对重复样本)检验所建立的PLS模型,以考察对木犀草苷定量校正模型的预测能力。预测集样品的制备方法、光谱测量条件、建模方法类似于校正集样品。图6中的虚心圈为校正集样品的散点,实心圈为预测集样品的散点,虚线为校正集样本的拟合曲线,实线为预测集样本的拟合曲线。由图6可见,模型的拟合值均匀分布在被拟合值点附近,两类样品偏PLS模型中预测浓度与参考浓度之间的拟合关系良好。在0.15~19.5 mg/L浓度范围内,测得预测集样品的相关系数为0.9765,预测均方根误差(Root mean square error of prediction,RMSEP)为0.9745 mg/L。木犀草苷预测集样本的回收率在84.7%~113.2%范围内,表明能够实现雪菊提取液中木犀草苷的富集,为近红外光谱的微量检测提供了可能。综上,雪菊提取液中木犀草苷经苯硼酸吸附剂后,即使存在有各种基体干扰,仍能被准确测定,因此可实现雪菊中木犀草苷的选择性检测。模型诊断一般用残差的结果进行判断,残差散点分布均匀,说明模型的性能良好[30,31];由图7的预测残差可见,各点残差值即预测响应值和实际响应值之差在0 mg/L上下呈对称分布,且具有恒定均匀的扩散,表明模型稳定,适于含量预测。因此,本研究成功建立了木犀草苷定量校正模型,并实现了对预测集样品含量的预测,结果良好,模型稳健有效;在复杂光谱背景下,经过CWT法对样品的近红外光谱进行处理和有效检测信息提取,可以准确、选择性地测定雪菊提取液中的木犀草苷含量。
4 结 论
采用温和方法制备了苯硼酸吸附剂,通过红外光谱进行了表征。将此吸附剂用于雪菊中木犀草苷的富集,與近红外光谱检测方法结合,实现了木犀草苷的定量测定。用苯硼酸吸附剂常温振荡吸附20 min,对木犀草苷的吸附效率可达到85.5%,吸附效果良好。将其用于雪菊提取液中木犀草苷的富集,采用近红外漫反射光谱方法对其中微量的木犀草苷进行定量分析,选择CWT对近红外光谱进行预处理,可达到最好的交叉验证结果。用PLSR法建立木犀草苷的定量校正模型,以29个样本作为预测集进行外部检验回归模型的准确性。在0.15~19.5 mg/L浓度范围内,预测集相关系数为0.9765,预测集中预测集样本的回收率在84.7%~113.2%范围内。结果表明,苯硼酸吸附剂提高了近红外光谱对木犀草苷的检测灵敏度,实现了雪菊提取溶液中微量木犀草苷的选择性检测,表明基于吸附富集策略与近红外光谱结合,采用化学计量学方法,可以实现药食两用植物中微量成分的分析和测定。
References
1 GUAN Ren-Wei,QU Yong-Sheng,GU Zheng-Wei,SHAO Xin,LIN Hui-Bin,LIN Jian-Qiang. Chinese Wild Plant Resources,2014,33(1): 1-3
管仁伟,曲永胜,顾正位,邵 新,林慧彬,林建强. 中国野生植物资源,2014,33(1): 1-3
2 Li Q L,Tian Z X,Wang M H,Kou J J,Wang C L,Rong X L,Li J,Xie X M,Pang X B. Int. Immunopharmacol.,2019,66: 309-316
3 Wang X Y,Yuan T,Yin N N,Ma X F,Zhang Z B,Zhu Z,Shaukat A,Deng G Z. Inflammation,2018,41(5): 1702-1716
4 Qing W X,Wang Y,Li H,Ma F Y,Zhu J H,Liu X H. AAPS Pharm. Sci. Tech.,2017,18(6): 2095-2101
5 ZHANG Gui-Lin,LIU Min,GE Hong-Juan,SONG Chun-Mei,WANG Dan,ZHANG Ping. Food Science and Technology,2018,43(6): 242-245
张贵林,刘 敏,葛红娟,宋春梅,王 丹,张 平. 食品科技,2018,43(6): 242-245
6 Zhang B,Nan T G,Xin J,Zhan Z L,Kang L P,Yuan Y,Wang B M,Huang L Q. J. Pharmaceut. Biomed.,2019,170: 83-88
7 Yang D Z,An Y Q,Jiang X L,Tang D Q,Gao Y Y,Zhao H T,Wu X W. Talanta,2011,85: 885-890
8 Zhou W,Tam K Y,Meng M X,Shan J J,Wang S C,Ju W Z,Cai B C,Di L Q. J. Chromatogr. A,2015,1376: 84-97
9 Feng S X,Li X H,Wang M M,Hao R,Li M M,Zhang L,Wang Z. J. Pharmaceut. Biomed.,2018,148: 205-213
10 Huang Y,Feng Y,Tang G Y,Li M Y,Zhang T T,Fillet M,Crommen J,Jiang Z J. J. Pharmaceut. Biomed.,2017,140: 384-391
11 WANG Shi-Fang,CUI Guang-Lu,FENG Xiao-Yuan,HAN Ping. Journal of Food Safety & Quality,2017,(12): 4602-4608
王世芳,崔广禄,冯晓元,韩 平. 食品安全质量检测学报,2017,(12): 4602-4608
12 GUO Yu-Fei,SU Yi,XU Qing-Yang. Letters in Biotechnology,2016,27(3): 391-395
郭宇飞,苏 毅,徐庆阳. 生物技术通讯,2016,27(3): 391-395
13 Xie Y,Zhou R R,Xie H L,Yu Y,Zhang S H,Zhao C X,Huang J H,Huang L Q. Int. J. Biol. Macromol.,2019,122: 1115-1119
14 Chen X Y,Sun X F,Hua H M,Yi Y,Li H L,Chen C. Spectrochim. Acta A,2019,221: 117169
15 YU Hui-Ling,ZHANG Miao,HOU Hong-Yi,ZHANG Yi-Zhuo. Spectroscopy and Spectral Analysis,2019,39(8): 2618-2623
于慧伶,张 淼,侯弘毅,张怡卓. 光谱学与光谱分析,2019,39(8): 2618-2623
