
Hypoxia and oxidative stress:The role of the anaerobic gut, the hepatic arterial buffer response and other defence mechanisms of the liver
2020-05-19 06:22SamapriyaPasanHewawasam
World Journal of Meta-Analysis 2020年2期
Samapriya Pasan Hewawasam
Samapriya Pasan Hewawasam, Department of Physiology, Faculty of Medicine, University of Ruhuna, Galle 80000, Sri Lanka
Abstract
Key words: Anaerobic gut lumen; Oxidative stress; Liver defences against oxidative stress; Hepatic arterial buffer response; Hypoxia; Liver function
INTRODUCTION
The gastrointestinal tract has evolved for the digestion of food and absorption of nutrients to provide energy and nutritional requirements for the body.In nature, food is scarce and in intermittent supply; however, the energy needs of the body for metabolic processes are continuous.Evolutionary mechanisms were needed to store energy and nutrients during times of surplus (feeding episodes) for utilization during episodes of scarcity (episodes of fasting).In the never-ending quest for food, an innumerable number of toxic xenobiotic molecules occurring in nature can enter the body with food.These toxins are generated as by-products of chemical processes for the synthesis of food or are present as contaminants or are generated in the putrification of food and pose a risk to the health and the life of the animal.
The liver, which develops from the liver diverticulum, has evolved as the main storage organ for energy substrates and as the main detoxifying organ for ingested toxic xenobiotics.The liver has enormous catalytic potential due to the enzymes present within hepatocytes, which generate large amounts of oxidative stress that is injurious to hepatocytes.
Similarly, oxidative stress is generated within the intestinal lumen due to the oxidative enzymes of intestinal bacteria, which could oxidize and therefore lead to the breakdown of energy substrates, such as glucose, resulting from the digestion of complex carbohydrates.
Due to the unique arrangements of the blood supply of the intestines and the liver,the intestinal lumen and hepatocytes operate in a relatively hypoxic microenvironment.This hypoxic microenvironment is an important defence mechanism against oxidative stress at these sites, as will be elaborated in this review.
Challenge of storing energy
Energy sources (mainly complex carbohydrates and lipids) are digested and absorbed mainly in the small intestine.Complex carbohydrates are enzymatically broken down,mainly to glucose, in the small intestine, absorbed into the portal vein and transported to the liver for storage.
With regard to the enzymatically catalysed oxidation of glucose, two main substrates for the reaction are oxygen and glucose, whereas the end products can differ depending on whether it is a complete or partial oxidation.As per the chemical kinetics of any chemical reaction, the rate-limiting factor for the oxidation of glucose would be the concentration of the substrate with the lowest concentration of the two substrates if all other factors that affect the rate of the reaction are constant.On the other hand, if concentrations of both substrates are high in a catalytic environment of enzymes, that would accelerate the rate of the reaction, leading to the consumption of both substrates.
Hence, according to Michaelis-Menten kinetics for oxidative phosphorylation(occurring intracellularly) and other oxidative reactions of glucose mediated by bacterial oxidative enzymes (occurring in the lumen of the gut), the oxidation of glucose is stimulated in a catalytic environment of enzymes in the presence of high glucose concentrations if the oxygen tension of the milieu is also high[1,2].This can pose a challenge for the preservation of glucose in the intestines themselves if the oxygen tension of the intestinal lumen is high, as in the intestinal lumen, a high concentration of glucose is achieved by the digestion of carbohydrates and by bacteria that possess enzymes that can oxidize glucose, leading to the breakdown of glucose even before it reaches the liver for storage as glycogen.
Although, for practical purposes, the small intestine is traditionally considered“sterile”, it contains up to 104microorganisms/mL of intestinal secretions[3].Therefore,evolutionary adaptations are needed to prevent the oxidation of glucose within the intestines.The evolutionary adaptation in the intestines to overcome this challenge was to make the lumen deeply hypoxic to the extent that the oxidation of energy substrates such as glucose are inhibited due to the lack of oxygen.The degree of hypoxia achieved in the intestinal lumen is so severe that the intestinal lumen is traditionally considered “anaerobic”, and only facultative anaerobes and obligate anaerobes colonize it[4].Friedmanet al[5]attributed the colonization of intestinal lumen by anaerobic bacteria to the consumption of luminal oxygen by facultative anaerobes and to other chemical reactions that utilize oxygen, which convert the lumen into a deeply hypoxic environment.These microorganisms consume oxygen that diffuses into the gut lumen from mucosal capillaries.The action of these microbes is prominent only in the colon, contributing substantially to deep hypoxia of the colon only, where the size of the biomass is considerably larger[5].
Friedmanet al[5]considered chemical reactions consuming oxygen (e.g., oxidative reactions such as lipid peroxidation, seen in germ-free mice) as one of the important mechanisms contributing to the generation of the anaerobic intestinal lumen,especially in the proximal small intestine.Although they considered chemical reactions consuming oxygen (e.g., lipid peroxidation) as an important mechanism contributing to the anaerobic gut lumen in the small intestine, these oxidative reactions can also be viewed as undesirable yet unavoidable reactions due to oxidative stress resulting in the breakdown of energy substrates.This undesirable lipid peroxidation in the small intestine has been minimized by another evolutionary adaptation to make the lumen of the intestine as hypoxic as possible, as described below.
In the small intestine, where the energy substrate concentrations are highest, the anaerobic (more strictly, deeply hypoxic) gut lumen is achieved by a remarkable evolutionary adaptation in the microvasculature, preventing the diffusion of oxygen into the gut lumen.In small intestinal villi, there are hairpin loop arrangements between arterioles and venules with multiple cross connections between them.Within the villi, afferent and efferent vessels are separated by a distance of only 20 μm[6,7].This arrangement allows for a countercurrent shunt in which oxygen carried into the villus is able to diffuse to the venule without being transported through the vascular circuit bound to red blood cells, thus reducing the oxygen content of blood delivered to the villous tip[7,8](Figure 1).
These adaptations help to maintain the lumen of the small and large intestine in a deeply hypoxic state, supporting the colonization of the small and large intestine with anaerobic bacteria and discouraging the growth of aerobic bacteria.Anaerobic fermenters of the gut oxidize glucose and other simple sugars to lactate, which can be salvaged in the liver with the help of the Cori cycle to re-synthesize glucose[9].If the luminal environment of the gut was conducive to aerobic bacteria that possess cytochromes and other oxidizing enzymes, they would oxidize glucose to CO2and water, leading to complete wastage of energy substrates.
There is a similar challenge in preventing the oxidation of these energy substrates by the action of oxidative enzymes present in high amounts in hepatocytes once they reach the liverviathe portal vein.In this catalytic environment, during the fed state,glucose is oxidized to pyruvate, then is fluxed into mitochondria along the concentration gradient and is oxidized at a rapid rate if the oxygen concentration in this milieu is also high.However, if the oxygen concentration (tension) of this environment is maintained at low levels, oxidative phosphorylation is inhibited due to hypoxia, and pyruvate will be reconverted to glucose in the cytoplasm.This glucose can then be stored as glycogen once the adenosine triphosphate (ATP),guanosine triphosphate and uridine triphosphate levels become sufficiently elevated as a result of anaerobic metabolism of glucose.
Multiple lines of evidence for the metabolic stress placed upon the liver for the oxidation of energy substrates exist.The liver is the largest contributor to the basal metabolic rate of 27% in the adult and is one of the four high-metabolic-rate organs[10,11].The contribution of the liver to basal thermogenesis and, more importantly, the contribution of the liver to diet-induced thermogenesis can be considered additional evidence for the stress placed upon the liver for the oxidation of energy substrates due to its enormous catalytic reserve[12-15].It has been shown that diet-induced thermogenesis is more prominent with meals containing medium-chain fatty acids that are absorbed into the portal vein than with meals containing longchain fatty acids that are absorbed into the lacteals and deliveredviathe intestinal lymphatics to the systemic circulation, bypassing oxidative metabolism in the liver[16].
Challenge of protecting the body from the toxic xenobiotics present in food
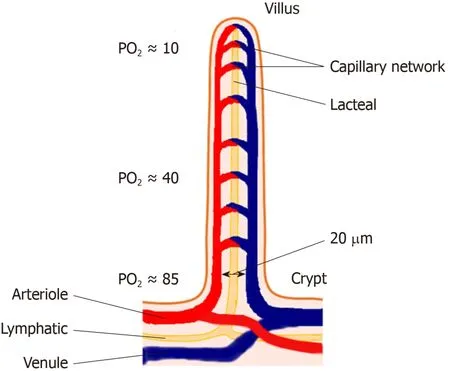
Figure 1 Schematic diagram of countercurrent blood flow within the small intestinal villus allowing diffusion of O2 from the arteriole to the venule, creating a luminal hypoxia from the crypt to the villous tip.
During digestion, the gastrointestinal tract is exposed to an innumerable number of naturally occurring toxic xenobiotics in food, of which some are lipid soluble and penetrate biological membranes with ease.The body is confronted with the challenge of protecting the organ systems, especially the central nervous system, from these toxic molecules through detoxification and excretion mechanisms.The liver,developing from the liver diverticulum of the embryonic gastrointestinal tract, which also drains the blood supply of the gastrointestinal tractviathe portal vein, has evolved as the organ for the detoxification of these substances prior to their release into the systemic circulation.The liver accomplishes this task mainly by the oxidative metabolism of these toxic substances within hepatocytes and converting them to polar, water-soluble substances (to which cell membranes are relatively impermeable).These toxic metabolites are either excreted in urine or conjugated with glucuronic acid and excreted back into the gastrointestinal tract with bile to be excreted with faeces (e.g., the excretion of haem in food as bilirubin diglucuronide).
The detoxification process of most toxic substances by the P450 enzyme system within the hepatocyte is achieved by binding these toxic molecules to P450 enzymes and subjecting them to controlled oxidation using oxygen, leading to less toxic end products[17,18].However, during this oxidative process, highly reactive free radicals and oxygen species are formed inside hepatocytes, which can react and damage the cytochrome P450 system itself as well as other cellular proteins, cell membrane constituents (lipid peroxidation) and genetic material, including DNA, leading to hepatocellular damage and necrosis.It has been shown that hepatocytes experience increased oxidative stress and suffer more cellular injury during the oxidative metabolism of toxic substances such as acetaminophen if the hepatocytes are exposed to higher oxygen tensions during the oxidative process[19].
Indirect evidence for the generation of increased amounts of reactive oxygen species and oxidative stress that damage the cytochrome P450 enzymes and other cellular elements comes from a study by Suleimanet al[20], where they showed reduced rat hepatocyte viability, gradual loss of cytochrome P450 and the loss of cellular respiration when hepatocytes are cultured under higher oxygen tensions.
It is well established that the drug-induced hepatotoxicity of halothane is caused by the formation of highly reactive trifluoroacetyl metabolites of halothane during oxidative metabolism by liver microsomal P450 enzymes[21].These trifluoroacetyl metabolites then react with hepatocyte proteins to form antigens, triggering immunological damage to hepatocytes.It has been shown that the formation of these trifluoroacetyl metabolites is increased by increasing oxygen tensions[21].This means that reactive metabolite and reactive oxygen species production is increased when the oxygen tension of the hepatocyte milieu is increased during the detoxification of xenobiotics.
To minimize this damage, the cytochrome P450 system of the liver has evolved to operate under highly controlled conditions where oxygen tension of the reactive environment is kept to a minimum, minimizing the generation of free radical and toxic intermediates while providing sufficient oxygen to produce adequate ATP to power the cellular processes.If the cytochrome P450 system is exposed to high oxygen tensions during this process, large quantities of highly reactive toxic intermediates and oxygen species are produced, leading to increased hepatocellular damage and sometimes fulminant hepatic failure.
DEFENCE MECHANISMS OF THE LIVER AGAINST OXIDATIVE STRESS
There are multiple protective mechanisms in the liver to circumvent oxidative stress.The role of the intracellular antioxidant glutathione in combating oxidative stress is extensively described in the literature and will not be discussed here[22].
Some of the most important protective mechanisms to circumvent oxidative stress in the liver, undescribed in the form conceptualized in this article until now, are the evolutionary adaptations to maintain a relatively hypoxic microenvironment within hepatic sinusoids to achieve a low oxygen tension inside hepatocytes.
Hepatic arterial buffer response as a defence against oxidative stress
The liver receives 75%-80% of its blood supply from the portal vein, which drains the partially deoxygenated venous blood from the spleen, stomach, small and large intestine, gallbladder and pancreas, while the hepatic artery accounts for the remaining 20%-25%, supplying the liver with well-oxygenated blood[23].Branches of the portal vein and hepatic artery supply the hepatic sinusoids, in which the mixed blood flows towards the hepatic venules at the centre of the hepatic lobules.These hepatic sinusoids are lined on their external surface by plates of hepatocytes, which modify the composition of the blood flowing in the sinusoids.
The functional unit of the liver is a matter of debate, and the concepts of the“hepatic acinus” and the “primary lobule”, which are based on the blood supply and share some features in common, have gained wide acceptance within the scientific community[24,25].The concept of the hepatic acinus by Rappaport in particular has gained popularity since it allows for the explanation of lesions such as bridging necrosis and fibrosis[26-28].Each acinus is at the end of a vascular stalk containing terminal branches of portal veins, hepatic arteries, and bile ducts.Blood flows from the centre of this functional unit through the hepatic sinusoids to the hepatic venules at the periphery of the acinus, and the blood flow in the hepatic sinusoids is classically described as periportal-to-perivenular blood flow.
This gives rise to a functional zonation within the hepatic acinus where the central portion of the acinus closest to the mixed inflow of blood from the portal vein and hepatic artery (zone 1) receives the blood with the highest oxygen concentration, the intermediate zone (zone 2) receives less oxygenated blood, and the peripheral zone(zone 3) closest to the hepatic venules receives the least oxygenated blood[29](Figure 2).
The mixed inflow of blood to the acinus from the branches of the portal vein and the hepatic arterioles is regulated in a manner where there is an inverse relationship between the hepatic arterial and the portal venous blood flow.The regulation of hepatic arterial blood flow inversely with portal venous blood flow is called the“hepatic arterial buffer response” (HABR) and is mediated by adenosine[23].According to the currently accepted hypothesis for the adenosine-mediated mechanism of the HABR proposed by Lauttet al[30], adenosine is produced by hepatic metabolism in an oxygen-independent manner and is released at a constant rate into fluid in the space of Mall that surrounds the hepatic resistance vessels and portal venules.The concentration of adenosine is regulated by washout into the portal vein and the hepatic artery.If portal venous blood flow is reduced, less adenosine is washed away from the space of Mall, leading to an increase in adenosine levels, producing dilation of the hepatic artery with a subsequent increase in hepatic arterial inflow.
Although the existence of the HABR and the “adenosine washout hypothesis” are widely accepted, the main deficiency of Lautt’s theory is the inability to explain the functional significance of the HABR.This deficiency is connected to the source of adenosine for the HABR in Lautt’s theorem.According to Lautt’s hypothesis,adenosine for the HABR is derived from the demethylation of Sadenosylhomocysteine, a reaction that is oxygen independent.According to Lautt,adenosine, which is produced in huge amounts by hepatocytes in response to hypoxia, is secreted into hepatic sinusoids, where the blood flows away from the hepatic resistance vessels; therefore, the hepatic resistance vessels are not exposed to or influenced by adenosine produced in this manner.Nevertheless, in his “1995 Ciba-Geigy Award Lecture”, Lautt admitted that the pathway of adenosine production is an unresolved aspect of his hypothesis, and in this lecture, he proposed the demethylation of S-adenosylhomocysteine as the source of adenosine for the HABR[31].This proposal has gained wide acceptance as the source of adenosine for the HABR since then.
Significance of adenosine washout
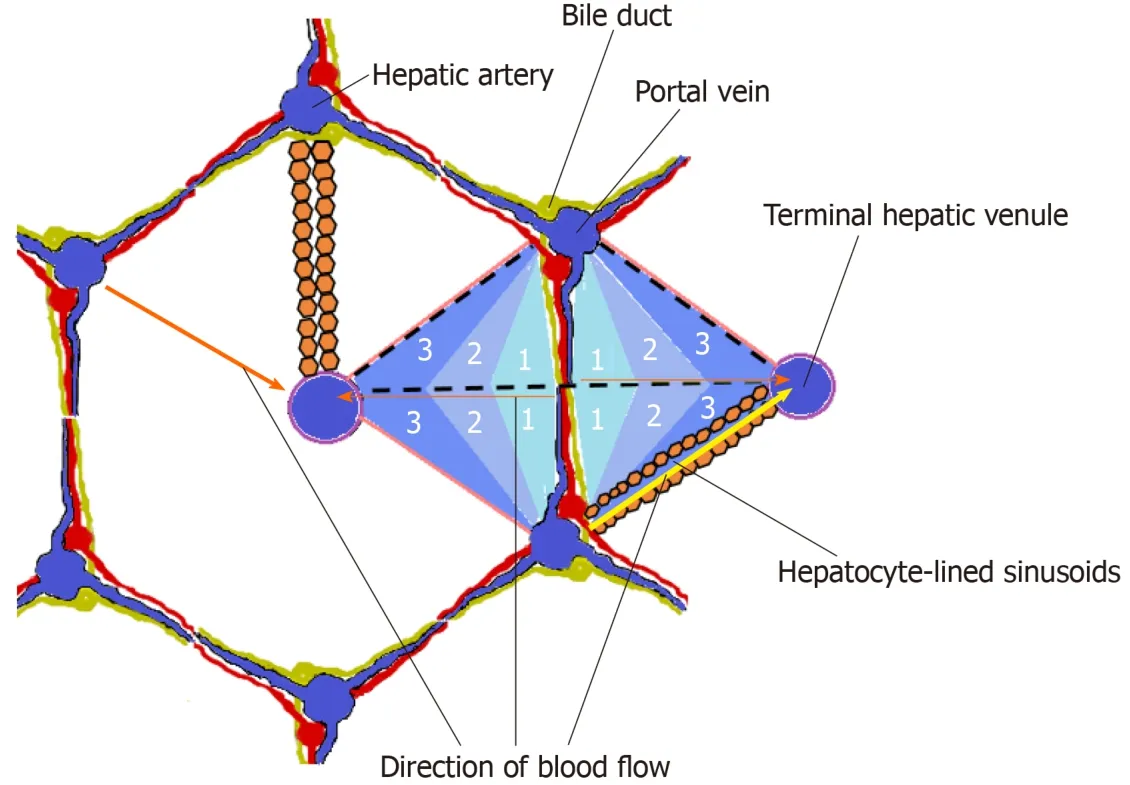
Figure 2 Schematic representation of a hepatic acinus within a conventional hepatic lobule.
Other researchers have subsequently shown the existence of hypoxia/anoxia-induced adenosine secretion by hepatocytes as an important mechanism of hepatic arterial vasodilatation during periods of increased hepatic hypoxia associated with portal flow reduction[32-36].Kurbelet al[36]described a model of dual circulation in hepatic acini, with hypoxia-regulated adenosine secretion that can drive the HABR, but this model has not reached wider acceptance despite its ability to support the functional significance of the HABR.
There is no rationale to assume that autoregulatory arteriolar dilatation due to hypoxia-mediated adenosine production seen in other organs like the heart will not occur only in the liver.Adenosine production can occur by the breakdown of adenine nucleotides or cyclic adenosine monophosphate, and these sources are directly linked to the energy status of the cells[31].In turn, the energy status of the cells is dependent on the oxidative phosphorylation of energy substrates such as glucose, which relies on an adequate oxygen supply.
The production of adenosine from adenine nucleotides, which occurs through multiple enzymatic steps, can be summarized as shown in Figure 3, which is in dynamic equilibrium.
This equilibrium is shifted towards ATP production under less hypoxic conditions that support oxidative phosphorylation and towards ATP depletion/adenosine production under critically hypoxic conditions.Thus, if the hypoxic microenvironment of the hepatocytes becomes critically hypoxic to the extent that it compromises oxidative phosphorylation with ATP depletion, adenosine production will be increased, producing hepatic arterial vasodilatation, with the correction of critical hypoxia.In other words, the characteristics of hepatic blood supply allow hepatocytes to operate under low oxygen tensions, which facilitates the controlled oxidation of toxic substances, minimizing the generation of harmful reactive intermediates and oxygen free radicals.This also minimizes the uncontrolled utilization of energy substrates, thus favouring the synthesis of storage polymers.
Enzymatic zonation as a defence against oxidative stress
Another protective mechanism of the liver against oxidative stress is the enzymatic zonation of different hepatocyte zones within the hepatic acinus, according to blood flow-dependent oxygen tensions, leading to metabolic zonation inside the hepatic acinus[37-39].Thus, metabolic processes requiring higher oxygen tensions/high CO2levels/high amounts of ATP, but that do not generate reactive intermediates and free radicals (e.g., oxidative energy metabolism, gluconeogenesis, urea synthesis,conversion of pyruvate to glycogen with the consumption of 6 ATP molecules), occur in the periportal area, which receives oxygen-rich blood.In contrast, reactions that require a lower oxygen tension for controlled oxidation occur in the perivenous zone,where the oxygen tension is low.Examples of such reactions include the controlled oxidation of glucose to glucuronic acid and glucuronidation, reductive reactions such as glycogen synthesisviaUDP glucose, glutamine formation and finally reactions that require the prevention of the uncontrolled oxidation of certain substrates to minimize the generation of reactive intermediates and oxygen free radicals (e.g., xenobiotic metabolismviathe microsomal cytochrome P450 system).This is achieved by the differential expression of enzymes among periportal and perivenous zones (called enzymatic zonation) and is probably achieved by an oxygen-mediated/dependent mechanism for gene expression in hepatocytes[40].

Figure 3 Production of adenosine from adenine nucleotides.
Perivenous (zone 3) zonation of the cytochrome P450 system confers a survival advantage by minimizing the generation of reactive intermediates and oxygen species during xenobiotic metabolism, thus minimizing hepatocellular damage.These perivenous hepatocytes may completely clear the blood of xenobiotic toxins during their first pass through the liver when the dose ingested is low.This advantage is not present when xenobiotic toxins are ingested in large doses or enter the bodyviathe parenteral route orviainhalation (e.g., halothane).
This enzymatic zonation according to the oxygen gradient within the hepatic acinus also leads to the manifestation of zone-specific patterns of liver disease.For instance,most xenobiotics (including the “cultural poison’’ ethanol) that are metabolized by the microsomal cytochrome P450 system, which is preferentially expressed in the perivenous area (zone 3), give rise to hepatocellular damage and necrosis in the perivenous area due to the generation of reactive intermediates and oxidative stress[41].Acetaminophen overdose also causes zone-3 necrosis when the highly reactive oxidative metabolite of acetaminophen, N-Acetyl-p-benzoquinone-imine (NAPQI),exhausts the capacity of the glutathione scavenger system to clear NAPQI, leading to covalent binding of NAPQI to cellular proteins[42].It is interesting to note that although N-acetylcysteine and methionine are successful in replenishing hepatocellular glutathione, most of the other antioxidants may not be able to penetrate into hepatocytes to scavenge oxygen free radicals and reactive intermediates.Halothane hepatotoxicity also causes zone 3 necrosis due to its metabolism by the microsomal cytochrome P450 system expressed preferentially by perivenous hepatocytes[43].It has been shown that microsomal cytochrome P450 inducers such as isoniazid, phenytoin, phenobarbital and alcohol can augment hepatocellular damage due to oxygen free radicals and reactive intermediates.
Most of the aetiologies that produce cirrhosis, such as alcohol, haemochromosis,Wilson disease and non-alcoholic steatohepatitis, lead to hepatocellular damage through disturbances they cause to the oxidative catalytic potential, and when the oxidative stress is overwhelming, many of these diseases show Fenton or Fenton-like reactions within the hepatocytes with the production of highly toxic hydroxyl free radicals, further perpetuating the liver damage[44-46].
Regenerative capacity of the liver as a defence mechanism against oxidative stress
Of all the vital organs in the body, the liver has the most remarkable regenerative capacity.It can regenerate to its original size after a 70% hepatectomy within approximately two weeks while preserving all liver functions and requires no more than two cycles of hepatocyte replication[47].More importantly, the liver can regenerate fully after massive hepatocyte necrosis, as occurs in toxin-mediated necrosis, such as acetaminophen hepatotoxicity or ischaemic necrosis[47,48].This is particularly important as reactive intermediates and oxygen free radicals can bind to genetic material within hepatocytes and cause apoptosis or hepatocellular necrosis.
The remarkable regenerative capacity of the liver, which differs from other organs of the body, is a property of the liver due to its hypoxic microenvironment.Although critical hypoxia leads to cell cycle arrest in most tissues, a lesser degree of hypoxia can also stimulate cellular proliferation in certain tissues and cells, such as embryonic tissues and cancer cells, as well as in hepatocytes, and this replication is orchestrated by signalling molecules, such as hypoxia inducible factors[49-52].
Hepatocytes can regenerate from a variety of precursors, namely, by the replication of existing hepatocytes, by the differentiation of oval cells (cells originating in the terminal branches of the bile ductular system and the canals of Hering) or from bone marrow cells under different circumstances with different triggering stimuli and different signalling molecules mediating such replication[47].Wanget al[53]recently identified a population of proliferating and self-renewing hepatocytes in centrilobular zone 3 adjacent to the central vein of the lobule in mice in the absence of any liver injury.These cells were shown to proliferate twice as fast as the other hepatocytes and replace more than one-third of the mouse liver lobule around the central vein during homeostatic renewal in approximately one year.On the other hand, there is another group of hepatocytes, called hybrid hepatocytes, in zone 1, near the portal triad,which undergo extensive proliferation and replenish liver mass after chronic hepatocyte-depleting injuries[48].Therefore, it can be concluded that different hepatocytes harbour different proliferative capacities.This seems to carry functional significance with regard to different triggers that invoke different patterns of hepatocellular damage and necrosis and for the restoration of hepatic mass and architecture or for the development of nodular regeneration (cirrhosis).
INSIGHTS INTO LIVER FUNCTION AND AVENUES FOR FUTURE RESEARCH
Strategic positioning of the spleen within the hepatic portal circulation is a fascinating evolutionary adaptation to the functions served by the liver and the spleen.Due to the highly stagnant and sluggish circulation within splenic sinusoids, there is increased oxygen extraction within splenic sinusoids compared to other tissues of the gastrointestinal tract.It is reasonable to expect that there is a significant contribution from this stagnant circulation in the spleen to the generation of portal venous hypoxia.The quantification of the contribution of the spleen to portal venous hypoxia is an area for potential research.Furthermore, because of the strategic position of the spleen, the largest contributor to bilirubin production in the body, within the hepatic portal venous circulation, most of the bilirubin that is produced by the spleen is cleared by the liver and excreted to the gut, thus preventing large-scale entry of bilirubin into the systemic circulation.The advantages of this arrangement are twofold.First, bilirubin is a lipid-soluble antioxidant which has complementary cytoprotective roles to glutathione and it can penetrate into cells to provide hepatocytes with an additional defence mechanism against oxidative stress and lipid peroxidation[54].Second, since a large fraction of the generated bilirubin is prevented from entering the systemic circulation, the neurotoxicity of bilirubin is minimized.
It may be worthwhile investigating the role of jejunal and ileal arcade vessels and their arterio-venous anastomoses in the maintenance of the anaerobic environment of the gut and the hypoxic environment of the liver.
There is a significant interaction between the gut and the liver through the portal vein and biliary secretions[55].Whether there is a role for this gut-liver crosstalk in the regulation of the portal venous flow and, if so, the nature of it and the molecules involved in it is another area for potential research.
Further research is needed to identify antioxidants that can penetrate into hepatocytes and neutralize oxygen free radicals/reactive intermediates as these would provide novel therapeutic options for diseases originating through oxidative stress.
One of the reasons for the partial success of artificial liver support systems seems to lie in their inability to replicate the anatomical relationship of the liver to the portal vein in its detoxification function as in normal physiology.It could be expected that artificial liver support systems would perform better if this problem could be addressed through the development of a mechanism to feed portal venous blood into the artificial liver support system rather than feeding systemic venous blood into it in patients with liver failure.
CONCLUSION
The liver, which can be seen as the “chemical smithy” of the body, has to work with“fire” (the catalytic potential) to serve its vital functions (Figure 4).Multiple evolutionary defence mechanisms are in place to defend the liver from the damaging effects of the oxidative stress generated by this enormous catalytic potential (Figure 5).A better understanding of these mechanisms will shed light on unravelling the mysteries of liver function and novel therapeutic options.
Figure 4 Liver as the “chemical smithy” of the body.
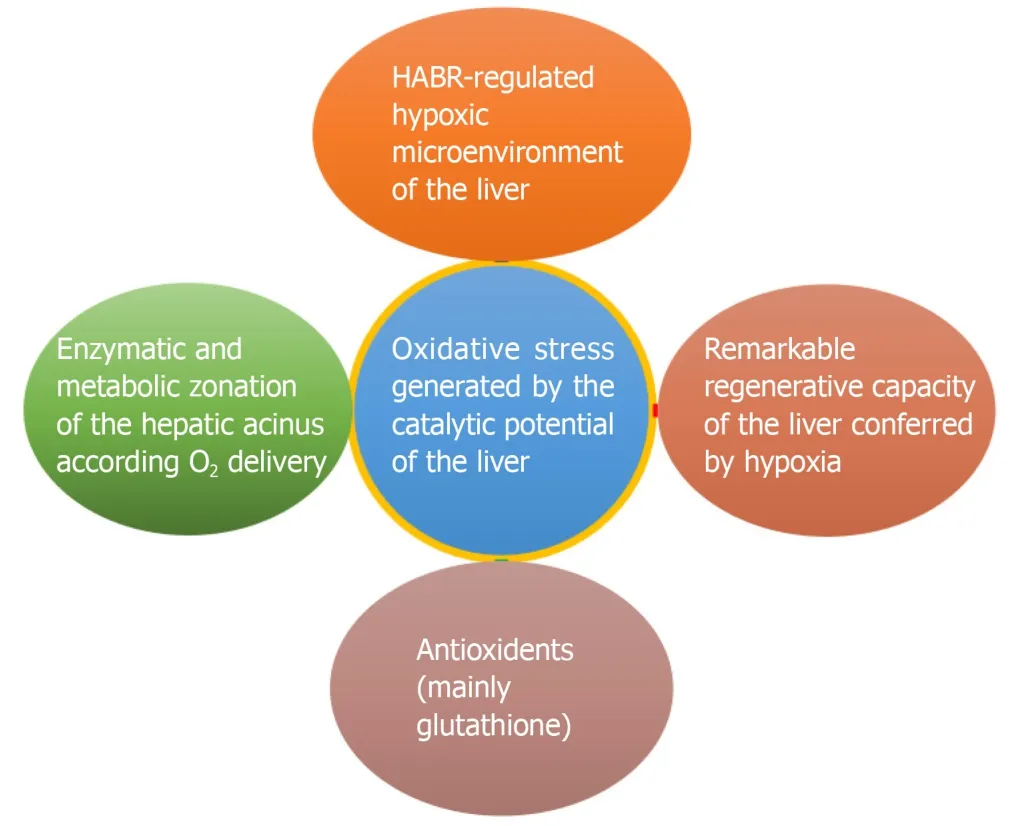
Figure 5 Protective adaptations of the liver against oxidative stress.
 World Journal of Meta-Analysis2020年2期
World Journal of Meta-Analysis2020年2期
- World Journal of Meta-Analysis的其它文章
- European Specialty Examination in Gastroenterology and Hepatology examination — improving education in gastroenterology and hepatology
- Probiotics in inflammatory bowel disease:Does it work?
- Pathological characterization of occult hepatitis B virus infection in hepatitis C virus-associated or non-alcoholic steatohepatitis-related hepatocellular carcinoma
- Helicobacter pylori and gastric cardia cancer:What do we know about their relationship?
- Treatment strategies and preventive methods for drug-resistant Helicobacter pylori infection
- Utility of gastrointestinal ultrasound in functional gastrointestinal disorders:A narrative review