
Glutamine transporters as pharmacological targets:From function to drug design
2020-05-15 09:35:34
Department of DiBEST (Biologia, Ecologia e Scienze della Terra), University of Calabria, Arcavacata di Rende (CS) 87036, Italy
Keywords: Glutamine transporters Drug delivery Pharmacokinetics Screening Proteoliposome
ABSTRACT Among the different targets of administered drugs,there are membrane transporters that play also a role in drug delivery and disposition.Moreover,drug-transporter interactions are responsible for off-target effects of drugs underlying their toxicity.The improvement of the drug design process is subjected to the identification of those membrane transporters mostly relevant for drug absorption,delivery and side effect production.A peculiar group of proteins with great relevance to pharmacology is constituted by the membrane transporters responsible for managing glutamine traffic in different body districts.The interest around glutamine metabolism lies in its physio-pathological role;glutamine is considered a conditionally essential amino acid because highly proliferative cells have an increased request of glutamine that cannot be satisfied only by endogenous synthesis.Then,glutamine transporters provide cells with this special nutrient.Among the glutamine transporters,SLC1A5,SLC6A14,SLC6A19,SLC7A5,SLC7A8 and some members of SLC38 family are the best characterized,so far,in both physiological and pathological conditions.Few 3D structures have been solved by CryoEM;other structural data on these transporters have been obtained by computational analysis.Interactions with drugs have been described for several transporters of this group.For some of them,the studies are at an advanced stage,for others,the studies are still in nuce and novel biochemical findings open intriguing perspectives.
1.Introduction
Membrane transporters are primary sites for drug interactions since they are located at the boundary between the intra end extracellular milieu [1–6].These interactions can occur at the level of absorbing or re-absorbing epithelia,that is intestine and kidney tubules,as well as at the level of other body districts which are in direct or indirect contact with the bloodstream.Unique cases are those of the bloodbrain barrier (BBB) and the placental barrier,responsible for controlling the flux of molecules to specific organs.In these districts,double-membrane barriers are present [7].From the described scenario it is evident that the absorption and the interaction of drugs with molecular cell systems are complex phenomena and are strongly influenced by the function or dysfunction of specific membrane transporters [8–10].Drugtransporter interactions are,then,expected to play key roles in human therapy [11,12]or,in other cases,to trigger side effects,due to the so-called off-target interactions [13].After some decades of investigations,it is now well accepted that membrane transporters must be taken into account for drug design to improve drug delivery and efficacy.In this respect,the International Transporter Consortium [14]has been founded for establishing:(i) which transporters must be taken into account for improving drug absorption;(ii) the suitable biotechnologies for assaying and screening drug-transporters interactions;(iii) the transporters to consider for off-target effects [15,16].The Society for Laboratory Automation and Screening (SLAS,https://www.slas.org) too,started to consider membrane transporters in drug discovery applications [17].The recent methodological advancements for studying transport proteins triggered an exponential growth of the research on membrane transporters and drug-transporter interactions [18–20].In this scenario,a great interest is deserved to a special group of membrane transporters:the glutamine transporters.Several reasons are at the basis of the growing interest towards this group of proteins,ranging from the improvement of basic knowledge to the involvement of the glutamine transport in crucial processes for cell life and their role in human pathologies.The last aspect opens new and very promising perspectives in exploiting these proteins as novel targets for human therapy.In this review,thestatusartisof this rapidly expanding field will be summarized.
2.Glutamine transporters relevant to the interactions with drugs:an overview
Some SLC (SoLuteCarrier) families include glutamine transporters whose role consists in contributing to the complex network of pathways underlying the glutamine metabolism.The main characteristics of glutamine transporters are the apparent redundancy and overlapping functions (Table 1) [21,22].This phenomenon is explained by the need of maintaining the glutamine homeostasis in the whole body.Glutamine is,indeed,the most abundant amino acid and plays a greater number of roles than the others.A schematic picture of the roles of glutamine as well as of the transporters handling glutamine is summarized in Fig.1,while a more detailed view of the glutamine pathways together with transporter expression profile under physiological conditions is available in previous papers[21,23,24].It has to be stressed that several roles of glutamine are tissue-specific,as an example,glutamine is synthesized in muscles or in neurons to scavenge ammonia but is degraded in the liver for urea production.The redundancy of transporters responsible for glutamine traffic in the human body may represent also a challenge in designing specific and safe drugs;however,this aspect may be afforded considering that the pattern of membrane transporter expression varies alot when comparing normal and pathological conditions and even among different pathologies.As an example,each cancer type is characterized by the expression of only one or a limited number of glutamine transporters that can be exploited for specific drug targeting.Given the pleiotropic role of glutamine,it is not a surprise that even if it is endogenously synthesized,it becomes conditionally essential under increased energy demand.In other words,highly proliferative cells are characterized by a strongly increased utilization of glutamine,together with glucose [11],for energy production bypassing or lowering the canonical mitochondrial production of ATP in the oxidative phosphorylation pathway.The use of glutamine as an energy source by cancer cells is also in line with the higher concentration of this amino acid in cells (from 2 to 20 mM) compared to that of the other amino acids [21,25].This metabolic rearrangement allows for an efficient ATP production at the substrate level by the TCA (Tricarboxylic Acid Cycle) enzyme succinyl-CoA synthase,besides by glycolysis.In this rewired conditions,the TCA cycle works in a truncated form in which malate is sequestered from the TCA to be exported in the cytosol.In the same scenario,glutamine carbon atoms are used for supporting anaplerotic reactions and for producing reducing equivalents,required for cell growth.It is worth noting that the described phenomenon is typical of cancer cells which are considered,indeed,glutamine addicted [26,27].In these cells,the endogenous synthesis of glutamine is not sufficient for supporting their high proliferation rate;as a consequence,a strong over-expression of some glutamine transporters is adopted by cancer cells [28].Therefore,these transporters represent a target for novel anticancer drugs focusing on cell metabolism,rather than on the mechanisms of proliferation as the currently employed chemotherapy.Over the years,efforts have been made to find the right application(s),based on these transporters,to increase the outcome of cancer treatment(s).A specific issue that has been taken into consideration,is the designing of non-covalent(competitive) and covalent (non-competitive) inhibitors(Fig.2).Indeed,the majority of transporter-based drugs are substrate-analogues and,therefore,act as competitive inhibitors with the consequent increase of the Km for the natural substrate (Fig.2 A).This approach,however,may result in a low efficacy of the treatment due to the displacement of the drug when the substrate concentration rises above the Km.The use of covalent inhibitors may overcome this problem since a covalently bound inhibitor cannot be displaced by the substrate (Fig.2 B).The setting up of this approach implies the knowledge of the structure of the transporter used as a target.In the following paragraphs,the major achievements on each transporter are summarized in terms of historical perspectives,basic functional information and interactions with drugs or potential drugs as principal applications in cancer treatment.
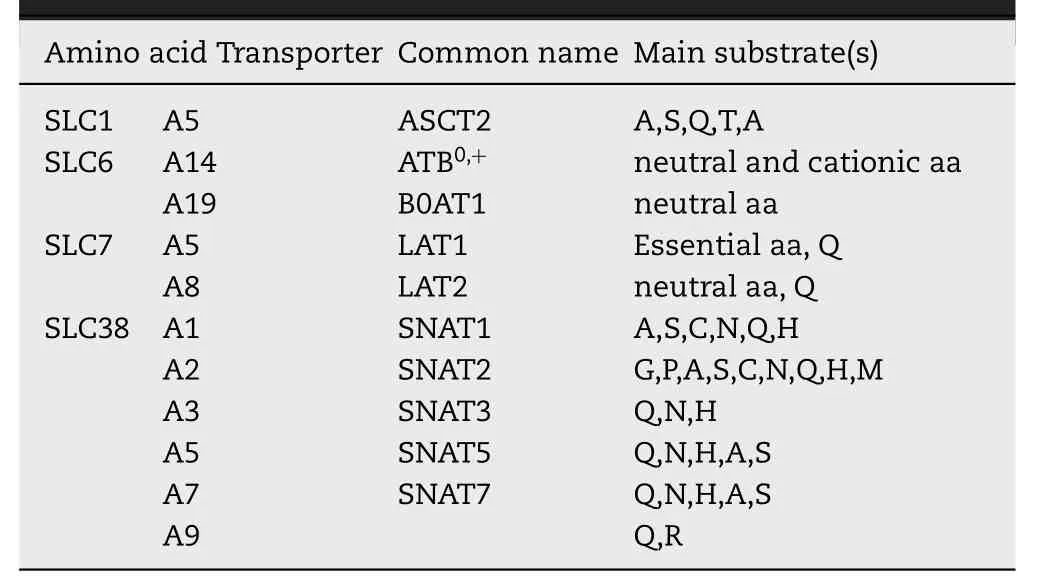
Table 1–The basic characteristics of membrane transporters that accept glutamine as a substrate.The SLC classification is reported with the common names;the major substrates of transporters are indicated.
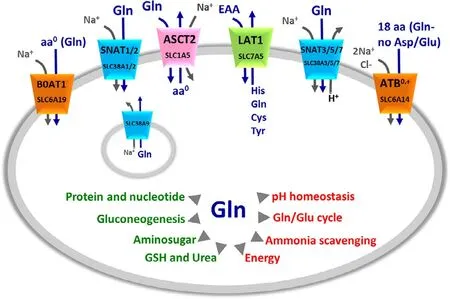
Fig.1–Glutamine membrane transporters and their mechanism of transport.The shape of the transporters is consistent with their asymmetry in the membrane.Membrane transporters are indicated by SLC classification and common names (as indicated in Table 1).The colors indicate the different families to which transporters belong:in orange SLC6 family,in green SLC7 family,in pink SLC1 family,in light blue SLC38 family.Lysosome localization of SLC38A9 is also indicated.Arrows indicate the direction of amino acids (blue) and ions (gray) fluxes.In the middle,schematic representation of Gln role in cells,the arrows indicate the main cell pathways in which Gln is involved(red) and the list of molecules synthesized from Gln (green).
2.1.The sodium-dependent neutral amino acid antiporter ASCT2 (SLC1A5)
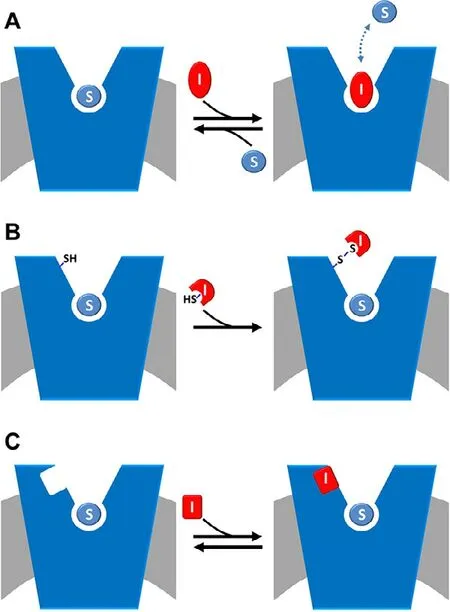
Fig.2–Schematic representation of inhibition kinetics.The membrane is in gray and a generic amino acid transporter is represented in blue and inserted in the membrane.(A)competitive mode is shown:the inhibitor,depicted in red(I),competes with the substrate (S),depicted in light blue,for the binding at the substrate binding site;the interaction is reversible as indicated as the double arrow.(B) covalent non-competitive mode is shown:the inhibitor,depicted in red (I) has a reactive group,exemplified by SH in the figure,that is able to covalently binds the protein in a site,alternative to the substrate binding site where also substrate is present (S,depicted in light blue).(C)non-covalent non-competitive mode is shown:the inhibitor,depicted in red (I) binds the protein in a site,alternative to the substrate binding site where also substrate is present (S,depicted in light blue).
ASCT2 (SLC1A5) is a plasma membrane protein [29]with a broad expression profile responsible for an obligatory exchange of neutral amino acid which is strictly Na+-dependent [29–32](Fig.1).Its function has been studied in different experimental models such as intact cells[33]and proteoliposomes reconstituted with the transporter extracted from rat kidney,human cell lines or produced by heterologous expression inP.pastoris[34–36].The common name of the protein is an acronym standing for AlanineSerineCysteineTransporter 2 (ASCT2),even though,it is now well assessed that glutamine is a major substrate[37,38]and that cysteine is not transported,but acts as an allosteric regulator [39].The transport of amino acids is functionally and kinetically asymmetrical,according to the profound structural differences between the extra and intracellular sides of the transporter [37,38].Under a physiological point of view,ASCT2 is involved in amino acid absorption in several tissues.A peculiar role of ASCT2 is that played in the glutamine/glutamate cycle between neurons and astrocytes.In this pathway,ASCT2 performs an interplay with other glutamine transporters belonging to SLC38 family(Fig.1) to remove the excess of glutamate from the synaptic cleft to reduce its toxicity [22,30].The regulation of transport activity is poorly understood.Physical interaction with the scaffold protein PDZK1,whose role is still not known,has been described bothinvitroandinvivo[38].
2.1.1.ApplicationsofASCT2indrugdesign
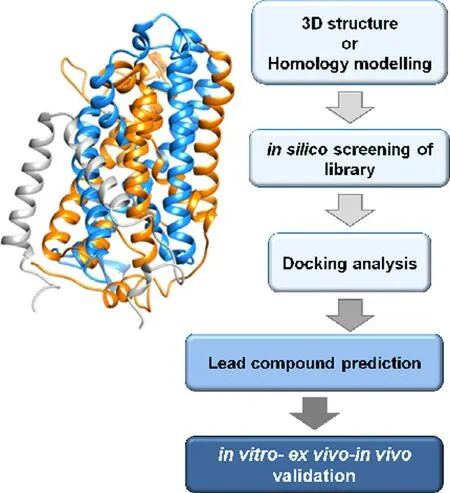
Fig.3–Work flow of best ligand prediction by in silico methodology.3D structure or homology structural model of a transporter (exemplified in the scheme) is required as a first step.Then,virtual high throughput screening of compounds from virtual libraries is realized followed by docking analysis for prediction of lead compounds.As a final step,the prediction is validated employing in vitro,ex vivo and in vivo methodologies.
Owing to the above-mentioned involvement of ASCT2 in glutamine absorption in cancer,in the last decades,efforts have been made to increase the knowledge on this transporter [40].Interestingly,virtually all human cancers are characterized by the over-expression of ASCT2 [28].This phenomenon is part of the well-known Warburg effect,characterized by the switch towards anaerobic glucose metabolism [26].These findings stimulated further investigations on new inhibitors of ASCT2 to be used as lead compounds for improving cancer drug design.These studies are performed either by synthesizing compounds whose structures harbor amino acid-like moieties or by predicting interactions of the protein with compounds downloaded from virtual chemical libraries to identify the best hits.This last strategy is based on the procedure of docking virtual chemical compounds to the 3D structure of the ASCT2 (or others) transporter (Fig.3),allowing high throughput virtual screening.Even though the 3D structure was not available at that time,a homology model has been obtained using specific approaches for improving the quality of the structure.The phylogenetic relationships among SLC1 family members and their prokaryotic homologs have been first analysed.Then,a model of ASCT2 has been generated performing a comparative analysis of the GltPh (glutamate transporter fromPyrococcushorikoshii) solved structure [41].The good quality of the produced model of ASCT2 has been then confirmed by the strong similarity with the recently solved structure of ASCT2 [37].The model has been then used for identifying several potential drugs.To evaluate the actual inhibition potency of each compound,drug-transporter interactions must be validated by biochemical assays.In this respect,the proteoliposome tool is very helpful for accurate measurement of IC50values and type of inhibition.It has to be highlighted that murine and human isoforms of ASCT2 exhibit an extensive difference in a local stretch of 31 amino acids.Even though this segment does not include the substrate-binding site,the difference may give variability in terms of interaction with molecules other than the substrate [38,42].As a further difference,the rat and the human isoforms harbor 16 and 8 cysteines,respectively [38].Taken together,this information indicates that the results collected with the murine isoform cannot be unequivocally transferred to the human ASCT2 without proper experimental testing.Notwithstanding,over the years the search for inhibitors have been conducted more or less in parallel with murine and human ASCT2.At first,a study on the rat ASCT2 proposed competitive inhibitors (Fig.2 A) based on glutamine derivatives.These compounds showed,however,relatively low affinity with IC50of about 500 μM [43]that is not suitable for specific targeting.Later more efficient inhibitors have been identified by combining docking with experimental validation (Fig.3):small side-chain amino acids with low hydrophobicity(and their derivatives) behaved as substrates of rat ASCT2,while molecules with large,aromatic side chains behave as inhibitors without being transported.Based on these criteria,a potent inhibitor,serine biphenyl-4-carboxylate,has been designed with a Ki of 30 μM.The results have been validated by measuring the transport function of rat ASCT2 following a non-physiological anion conductance in intact cells which is an indirect measurement of the transporter function [44].Later on,new molecules have been designed as inhibitors of the rat ASCT2 and validation has been performed as anion current or as radiolabeled glutamine uptake measurements in intact cells [45].It is worth noting that,in this case,the compounds have been designed based on proline structure,even though proline is not an ASCT2 substrate.Surprisingly,some of these compounds activate the transport,while others inhibit it.The best activator revealed to be a cis-3-hydroxyproline derivative while the inhibitor is a larger fluoro-benzylproline-derivative.These compounds,even if designed on rat ASCT2,showed cytotoxic effects also on human melanoma cell line [45].Moving from this indication,the benzyl proline derivatives constituted the scaffold for designing more efficient inhibitors specific for the human ASCT2 and a Ki in the micromolar range has been measured in human cells for these compounds [46].In another study,a competitive inhibitor of human ASCT2 has been identified starting from a previous screening conducted on 2-amino-4-bis (aryloxybenzyl)aminobutanoic acid derivatives [47].The lead compound,V-9302,reached the preclinical phase afterin silicoprediction andinvitroassays.Mice harbouring a patientderived xenograft (PDX) tumor revealed sensitive to the treatment with V-9302 that could reduce the tumor size.The molecular mechanism of inhibition of V-9302 is linked to a decrease of mTOR activity,consistent with lowered glutamine transport and metabolism and induction of autophagy and oxidative stress.Even though V-9302 is the first compound for which aninvivoeffect has been described,the efficacy and the specificity of this inhibitor are still under debate and some studies showed that the same compound can target also other amino acid transporters in cancer cells acting by a general mechanism on the amino acid metabolism [48,49].
An alternative approach to the described substrate analogues is that of covalent inhibitors based on chemically targeting the protein of interest (Fig.2 B).This approach would avoid the pitfall of displacement of the substrate analogues by the endogenous amino acids whose concentrations may increase in cancer cells [20].This strategy,collectively known as targeted covalent inhibitors (TCIs) [50],has been employed for the rat ASCT2 exploiting the presence of cysteine residues which are sensitive to several SH-reagents [51].Then,thiol reacting compounds based on a dithiazole moiety have been designed with IC50in the micromolar range,measured in proteoliposomes.The dithiazole ring is able to react with a thiol residue of cysteine forming a stable mixed disulphide (or a trisuphide) with the protein [52].The possibility of targeting ASCT2 with specific inhibitors with low side effects is indicated by some studies conducted on mice model in which the ASCT2 gene has been knocked out.In this condition,it has been demonstrated that the absence of ASCT2 does not affect the general cell metabolism,growth and proliferation.On the contrary,the KO of ASCT2 negatively affects the growth of cancer cells in which the transporter is overexpressed [53,54].Importantly,the structure of human ASCT2 has been recently solved [37]opening new perspectives in pharmaceutical studies related to cancer but also to the other pathologies in which ASCT2 is involved.The novel structure will allow performing more suitable high throughput virtual screening accelerating the identification of potent inhibitors of the transporter.
2.2.The sodium/chloride dependent broad amino acid transporter ATB 0,+ (SLC6A14)
ATB0,+(SLC6A14) is expressed in colon,lung [55],eye [56]and mammary gland [57](Table 1).This transporter recognizes glutamine together with all the other amino acids except for glutamate and aspartate [58].Transport is dependent on Na+and Cl−transmembrane gradients (Fig.1) and is sensitive to membrane potential [57].Interestingly,ATB0,+also recognizes carnitine as a low-affinity substrate.The relationships with glutamine metabolism are indirectly demonstrated by its overexpression in human cancers,thus representing a potential target of cancer therapy [59,60].
2.2.1.ApplicationofATB0,+indrugdesign
Few are the studies on the interaction with drugs with ATB0,+.This is also due to the scarce functional and structural knowledge of this transporter.Very recently,a novel computational approach has been used for predicting the 3D structure of ATB0,+allowing a more refined structural model of the transporter with respect to previous predictions[61].This represents the first step towards the application of the transporter in virtual screening and,hence,will give a further drive in designing possible drugs as ATB0,+inhibitors.So far,methyl-DL-tryptophan or BCH (2-aminobicyclo-(2,2,1)-heptane-2-carboxylic acid) have been exploited as chemical scaffolds for drug design based on substrate analogues(Fig.2 A and Fig.3) [58].These studies represent good starting points for further improvements in drug design using ATB0,+as a target.Interestingly,ATB0,+expression and function have been associated to cystic fibrosis even if,in this case,the role of ATB0,+is mostly linked to arginine transport and anion conductance rather than the transport of glutamine.Arginine is,indeed,responsible for nitric oxide production and this seems to be particularly relevant in CFTR mutant mice which are susceptible to ATB0,+KO,while WT mice are not extremely sensitive to the absence of this transporter in terms of overall metabolic changes [62].
2.3.The sodium-dependent broad amino acid transporter B0AT1 (SLC6A19)
B0AT1 (SLC6A19) is localized mainly in kidney and intestine[63–65].It is specific for neutral amino acids,among which glutamine,with half-saturation constants in the micromolar range,while basic and acidic amino acids are not substrate of the transporter (Table 1).Function and regulation have been studied in cells andinvitromodels:transport is strictly Na+-dependent (Fig.1) as expected for a transporter belonging to the SLC6 (Na+-dependent neurotransmitter family).The transport mediated by B0AT1 is electrogenic,with a 1:1 stoichiometry between the amino acid and Na+[66].It can be speculated thatinvivoB0AT1 is the major responsible for glutamine absorption in the intestine [65,67,68].The expression of B0AT1 is regulated by the Janus kinase 2 that stimulates its expression at the cell membrane [69],while leptin is a negative regulator.The localization of B0AT1 at the plasma membrane is regulated by ACE2 and by collectrin,a non-peptidase homologue of ACE2,in intestine and kidney,respectively [70].Collectrin has not any role in the intrinsic transport function [66];while the role of ACE2 in transport has not been clarified yet,even if it is plausible that the peptidase activity of ACE2 serves as a source of substrates for B0AT1[71].Despite its role in glutamine absorption and reabsorption,no variation of B0AT1 expression in cancer cells has been reported,so far.
2.3.1.ApplicationofB0AT1indrugdesign
The link of B0AT1 with human pathology relies on the mutation of the coding gene,which is causative of the Hartnup disease (OMIM 234500).The 3D structure of B0AT1 is still not solved.A homology model has been built using the structure of the bacterial transporter LeuT as template[72].Structure/function relationship studies have been performed in proteoliposomes:B0AT1 harbours two motifs responsible for metal binding,i.e.,CXXC and a CXXXC.These two motifs are at the basis of the inhibition exerted by heavy metals which have been demonstrated using proteoliposomes harbouring the rat protein.This interaction may represent one of the mechanisms of toxicity of heavy metals [66].The application of B0AT1 in drug design starts with discovering the interaction of the transporter with the commonly used drug nimesulide.The interaction has been characterized by combining inhibitor studies with kinetics and computational analysis.Very interestingly,molecular docking of nimesulide suggested that the drug binds to a site different from that of substrate binding located in a more external position producing a steric hindrance on the translocation path (Fig.2 C).This phenomenon has been displayed as a non-competitive inhibition when analysed by inhibition kinetics (Fig.2 C).The test of nimesulide analogues highlighted that minimal structural variations lead to a strong decrease of affinity revealing that the interaction with nimesulide is highly specific [72].Interestingly,the IC50for nimesulide is below 30 μM,considered as the threshold for predicting off-site interactions [73].Some of the side effects reported for nimesulide are similar,even though of lower intensity,to those of the Hartnup disease caused by B0AT1 defects [65].The side effects of nimesulide such as skin rush may rely on the reduction of absorption of some essential amino acids,such as tryptophan,triggering an impairment of niacin biosynthesis.Interestingly,a link of B0AT1 with fat-induced obesity has been recently proposed.It has been shown that disruption of the B0AT1 encoding gene in mice,induced a greater glycaemic control and resistance to obesity induced by high-fat diet.These data suggested that the pharmacological inhibition of B0AT1 could induce similar benefits in type 2 diabetes and obese patients.Thus,after nimesulide,additional inhibitors have been identified,such as benzotropine.These molecules decreased amino acid absorption in the organ model even though they exhibited an IC50higher (lower affinity) than that for nimesulide [74].Another inhibitor has been identified using a two functional cell-based assay employing fluorescence imaging methodology together with isotope-labelled amino acid transport.The identified compound,cinromide,is an amino acid analogue that is also transported by B0AT1,behaving thus as a competitive inhibitor (Fig.2 A) [75].
2.4.The histidine/large neutral amino acid antiporter LAT1 (SLC7A5)
L-type Amino acid Transporter 1 (LAT1) belongs to the large SLC7 family including two subgroups:the first includes cationic amino acid transporters;the second subgroup includes light subunits of heterodimeric amino acid transporters,among which LAT1 (SLC7A5) and LAT2 (SLC7A8)[76].
LAT1 is mainly expressed in testis,bone marrow,brain and placenta [76,77].In polarized epithelia,LAT1 protein shows a sub localization in basolateral membranes [76,78],while in BBB,this protein is localized also on the apical membrane [79].In the placenta,it is localized on both maternal and foetal surfaces of syncytiotrophoblasts [80].Intracellular localization of LAT1 has also been reported in the lysosomal membrane[81].In the plasma membrane,LAT1 forms a heterodimer with the glycoprotein CD98 (4F2hc,SLC3A2) by the formation of a disulfide between two conserved cysteine residues.From studies conducted in proteoliposomes harbouring the recombinant human protein,it has been demonstrated that CD98 has no role in the intrinsic transport function of LAT1[82].LAT1 is a Na+-independent obligatory exchanger of almost all the essential amino acids with a high preference towards histidine (Table 1) [83].Indeed,in knockout mice,His(histidine) accumulates in the brain demonstrating that it is the main efflux substrate in the brain [84].The role of LAT1 as glutamine transporter has been proposed in the past linking this activity to the high expression of this protein in human cancer [28,40].Later,it has been demonstrated that glutamine is a low-affinity substrate of LAT1 (Fig.1).Therefore,the role of LAT1 in glutamine traffic in cancer has been smoothened suggesting that this protein may be required by cancer cells for supplying essential amino acids [22].
2.4.1.ApplicationofLAT1indrugdesign
As in the case of ASCT2,the over-expression of LAT1 in cancers renders this transporter an exciting druggable target.Several reports dealt with chemical reagents designed as inhibitors of LAT1 for chemically knocking out the transporter in cancer cells following the common strategy depicted in Fig.3 .
As in other cases,two main types of inhibitors are the object of active research that is,competitive and noncompetitive inhibitors,proposed by virtual screening of drug libraries together with validationinvitroand/orexvivomodels (Figs.2 and 3).Within the first group,the amino acid analogue BCH,a well-known LAT1 inhibitor,has been firstly proposed as an anticancer drug [85].Other examples are phenylalanine,tyrosine and tryptophan analogues which showed some efficacy [86,87],as well as triiodothyronine(T3) analogues [88].Inhibitors based on hydroxamic acids conjugated to substrates of LAT1 have been also designed[89]with IC50ranging from 1 μM to more than 300 μM.The most effective competitive inhibitor so far designed is the tyrosine analogue JPH203.This inhibitor shows antitumor activityinvitroand in a mouse model of HT-29 tumours[90–92]as well as in other types of cancers [93–97].Very recently,JPH203 has been shown to markedly inhibit the proliferation of Anaplastic Thyroid Cancer (ATC) and to decrease the size of xenograft models [98].This represents one of most advanced result in the application of a transporter to drug design and cancer treatment.An approach based on computational and proteoliposome assay allowed identifying potent competitive inhibitors (Fig.2 A) based on the typical amino acid scaffold;a set of 30 compounds have been tested and two of them showed IC50in the low micromolar range [99].Very recently,a computational analysis has been conducted and several halogen-containing L -phenylalaninebased ligands have been identified displaying high affinity and high selectivity for LAT1;however,these data are stillinsilicoand need further experimental validation [100].Besides competitive,also non-competitive inhibitors have been designed moving from structure/function relationships studies (Fig.2B and 2C).Two cysteine residues,i.e.,C335 and C407,have been identified within the substrate-binding site of LAT1 [101].This finding suggested the strategy of designing covalent inhibitors able to react with the thiol groups of the cysteine leading to irreversible inhibition (Fig.2 B).Therefore,compounds harbouring a dithiazole group have been synthesized and tested for inhibition.Some of the compounds led to stable inhibition with a sub-micromolar IC50value measured in proteoliposomes.The molecular mechanism of such interaction has been demonstrated by employing site-directed cysteine mutants that lost the reactivity towards the compounds.The effect on cancer cell death has been also assessed [102].Interestingly,the structure of dithiazoles is“histidine-like”in line with minimal requirements for a molecule to interact with the substratebinding site of LAT1.The His-like property has been recently exploited also by another group that designed 1,2,3-triazolyl analogues [103].This interesting application links the ability of a molecule to reach the substrate binding site with the capacity to form stable covalent binding.This strategy is expected to increase a lot the potency and efficacy of an inhibitor.The task of designing novel drugs will benefit from the recent solution of the 3D structure of human LAT1 in complex with CD98 [104].Indeed,the screening of virtual library using the actual 3D structure of the transporters will give more reliable information on the affinity of the compounds identified as good interactors of the transport protein.Moreover,as in the case of ASCT2,the comparison of the previously predicted structures with the actual 3D structure of LAT1 [105]could give information on the validity of the previously identified compounds.
2.5.The neutral amino acids antiporter LAT2 (SLC7A8)
L-type Amino acid Transporter 2 (LAT2) belongs to the large SLC7 transporter family,as LAT1.
LAT2 (SLC7A8) is widely expressed and,in polarized epithelial cells,it is mainly localized at the basolateral membranes.LAT2 works as a heterodimer coupled to the glycoprotein SLC3A2,known as 4F2hc or CD98 [106,107].The interaction between the two proteins occurs by a disulphide between two conserved cysteine residues,as in the case of LAT1 [76].The transport mechanism of LAT2 is the same as LAT1 even though the substrate specificity is different.LAT2 is a transporter of small neutral amino acids,including glutamine,with Km in the millimolar range (Table 1) and is inhibited by the non-metabolizable amino acid analogue BCH[108].
A role in cancer growth has been demonstrated for LAT2 that regulates glutamine-dependent activation of mTOR,promoting glycolysis.This molecular mechanism activates chemo-resistance [109].Therefore,LAT2 represents another potential target in pharmacology,even though no application on interactions with drugs have been described so far for this protein.
2.6.The glutamine transporter SLC38A9
SLC38A9 is a peculiar amino acid transporter belonging to the SLC38 family that includes 11 members,some of which are well-studied,others are still orphans (Fig.1) [110].Historically,these transporters are known as SNATs and have been classified in systems A and N,following the features of transport modes and substrate specificity.In particular,System A includes SNAT 1 and 2 (SLC38A1/A2) that mediate the Na+-dependent uniport of small neutral amino acids and are inhibited by MeAIB (MethylAminoIsoButyric acid).System N includes SNAT3,5 and 7 (SLC38A3/A5/A7) that mediate the uptake of glutamine,histidine and asparagine with a transport mode dependent on both Na+and H+.Despite the importance of these transporters in glutamine disposition,interactions with drugs are not yet investigated.Among the orphan transporters,the SLC38A9 has been recently deorphanized.This protein has been defined“transceptor”,i.e.a transporter with a receptor function [111–114].It is localized in the lysosomal membrane (Fig.1) and at the difference with other canonical transporters,the transceptor SLC38A9 catalyses a glutamine efflux at a relatively low rate.The efflux of glutamine is allosterically regulated by intralysosomal arginine with consequent activation of SLC38A9 receptor function.This consists of the mTORC1 recruitment to the lysosomal membrane and following activation of the mTOR pathway [111–114].Given the ability to interact with one of the master protein in cell growth,SLC38A9 represents one of the most promising targets in the future drug design perspective.However,no application is so far described for this transporter.
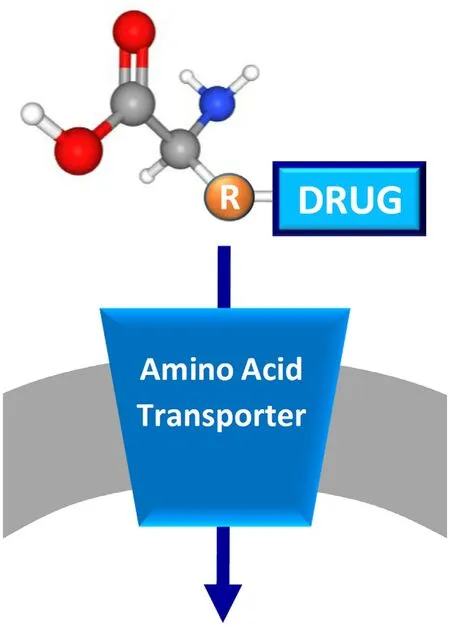
Fig.4–Schematic representation of prodrug approach.The membrane is in gray and a generic amino acid transporter is represented in blue and inserted in the membrane.The prodrug is depicted by a generic amino acid scaffold with the coupled drug bound to the side chain (R).
3.The prodrug approach:exploiting ATB 0,+and LAT1 transporters
The localization of membrane transporters at the boundary with the extracellular environment makes these proteins relevant for mediating drug delivery and not only as drug targets.An interesting strategy is famous as the prodrug approach designed to increase the efficacy of a pharmaceutical compound by improving its pharmacokinetics,i.e.the uptake in the target cell.In particular,the prodrug can be any amino acid recognized by a specific transporter,conjugated with the pharmacological compound (Fig.4) [115].This approach is used on the one hand to enhance drug uptake by intestinal absorption as an example [116,117],on the other to reduce drug affinity towards efflux transporters and,thus,to circumvent the multi-drug resistance typical of some pathologies such ascancer [118].The employment of amino acids has usually the advantage of increasing the water solubility of the prodrug through the presence of ionized carboxylate or amino groups[115].Furthermore,the use of amino acids is suitable due to the redundancy of the amino acid transporters such as those handling glutamine (Table 1).Among these proteins,ATB0,+(SLC6A14) and LAT1 (SLC7A5) have been,so far,the most employed in the prodrug approach,for different reasons(Table 2).In the case of ATB0,+,the choice is due to its feature of high capacity transporter and to its localization in the intestine that allows the rapid uptake of prodrugs.In the case of LAT1,the choice is mainly due to its expression in the blood brain barrier,a body district in which the flux of drugs is very limited.Therefore,the possibility of conjugating a drug with a LAT1 substrate increases a lot the possibility of targeting the brain to treat neurological disease,such as Alzheimer’s and Parkinson’s Diseases [89,119–123].Lastly,the over-expression of ATB0,+and LAT1 in several human cancers makes the prodrug approach another important possibility for improving the treatment of this multifaceted disease.Examples of prodrugs are the esters of acyclovir and ganciclovir with an alpha-carboxyl group of neutral amino acids that become substrates of ATB0,+[124].Furthermore,serine and threonine are also used for delivering coupled drugs into cells via ATB0,+[56].Anticancer drugs designed as arginine derivatives and as serine derivatives of irinotecan have been employed as substrates of ATB0,+in cervix and in breast cancer,respectively [125,126].Notably,considering that ATB0,+mediates a low efficacy transport of carnitine,the butyrate ester butyryl-L -carnitine revealed to be a prodrug to treat gut inflammation [127].Concerning LAT1,several reports have dealt with the use of natural amino acids as pro-moiety of L -DOPA,7-chlorokynurenic acid and ketoprofen ([83,119,128]and refs herein).Furthermore,the protease inhibitors saquinavir,indinavir and nelfinavir,largely used to treat HIV infections,have been conjugated to phenylalanine and leucine for increasing absorption through LAT1 [129].Interestingly,also prodrugs with chemical structures different from amino acids have been designed to be transported by LAT1 in brain,as dopamine derivatives[130].In prostate cancer,quinidine derivatives linked to valine and isoleucine cross cell membranes via LAT1 transporter[131].Prodrug approach,based on LAT1 substrate,has been attempted also to treat brain cancers which are normally characterized by high resistance to chemotherapy [132].Given the mentioned substrate redundancy for amino acid transporters,is not trivial that the prodrug approach will receive further advancements and that,other amino acid transporters,besides ATB0,+and LAT1 will be exploited as a target of pro-drug approach.A list of the prodrugs is reported in Table 2 .
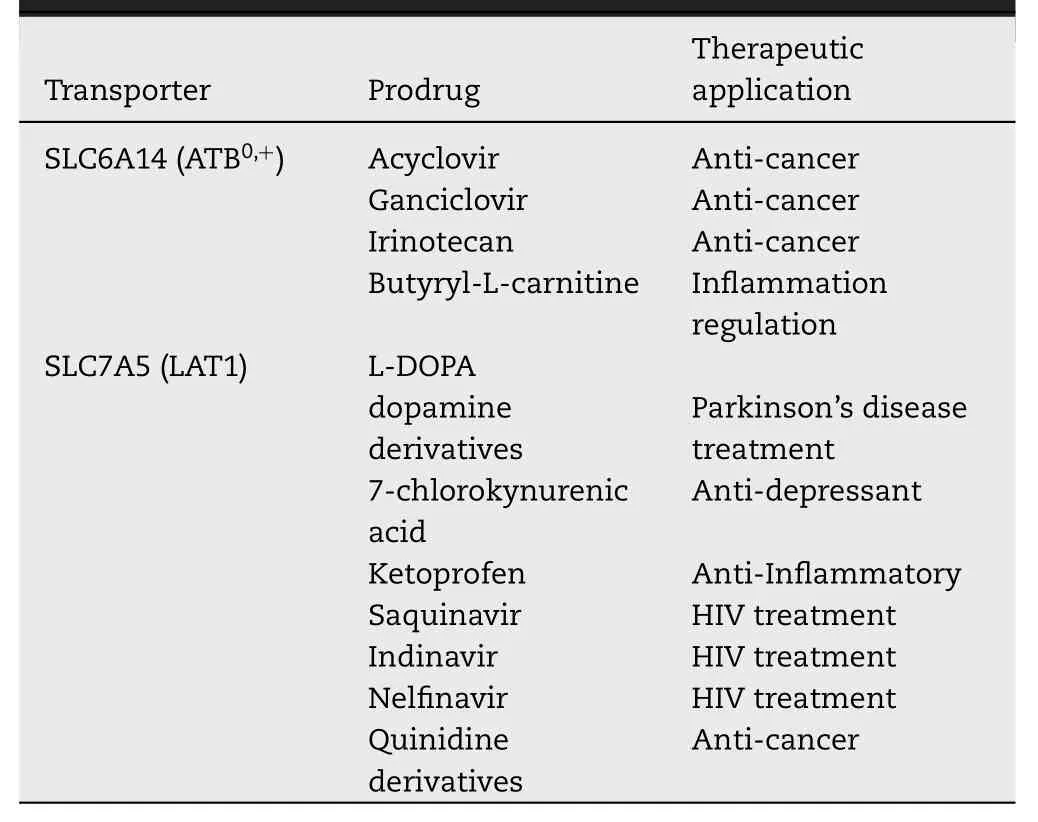
Table 2–Prodrugs interacting with SLC6A14 and SLC7A5.
4.Nanoparticle approach for drug delivery:exploiting glutamine transporters
A more and more frequently used approach for improving drug delivery is that of using nanoparticles as a vector of drugs,that can be associated or covalently bound to such a nanomaterial.Some approaches have been already attempted in the case of transporters and,in particular,transporters dealt with in this review.As an example,glutamate-conjugate paclitaxel nanoparticles or L -dopa conjugated nanoparticles have been exploited to increase the absorption of these drugs via LAT1 [133].The same transporter has also been exploited to increase the absorption of amino acid-modified drug associates-nanoparticles in the tumours that highlyexpressed LAT1 [134].Another transporter employed in nanoparticle strategy for drug delivery is ATB0,+.However,in this case,the protein has been considered for its capacity to transport L -carnitine besides amino acids.In particular,L -carnitine conjugated nanoparticles have been shown to increase the absorption of some drugs such as 5-fluorouracil[135].It has to be stressed that the described data represent preliminary investigations and the actual transport processes via the amino acid transporters are still not demonstrated.Anyway,this strategy represents an important starting point for alternative treatments of tumours not responding to the conventional therapies.
5.Conclusion
As described in the previous paragraphs,the interest around membrane transporters and their role in pharmacology increased a lot in the last years as testified by the launch of the International Transporter Consortium and by the FDA (Food and Drug Administration) recommendations and guidelines for studying interactions of transporters with drugs [16].The involvement of these proteins in common diseases,such as cancer,diabetes and neurological disorders,renders glutamine transporters hot spot targets for improving human therapy.Moreover,the location of specific glutamine transporters in specialized districts of the human body makes these transporters a vehicle for delivering the continuously increasing number of prodrugs.Notwithstanding the importance of the glutamine transporters and their relevance to human health,the interactions with drugs are still not accomplished and a big effort is required to obtain an exhaustive description of the expected involvement of the glutamine transporters in drug delivery and interaction.
Conflicts of interest
The authors report no conflicts of interest.The authors alone are responsible for the content and writing of this article.
Acknowledgments
This work was supported by PON (Programma Operativo Nazionale) Project No.01_00937 and by PRIN (Progetti di Ricerca di Rilevante Interesse Nazionale) Project No 2017PAB8EM to CI granted by MIUR (Ministry of Education,University and Research) Italy and by PRIN (Progetti di Ricerca di Rilevante Interesse Nazionale) Project No 2017PAB8EM.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.ajps.2020.02.005 .
 Asian Journal of Pharmacentical Sciences2020年2期
Asian Journal of Pharmacentical Sciences2020年2期
- Asian Journal of Pharmacentical Sciences的其它文章
- Special topic:Emerging role of transporters in drug interaction and delivery
- The influence of genetic polymorphisms in drug metabolism enzymes and transporters on the pharmacokinetics of different fluvastatin formulations
- Organic anion transporters also mediate the drug–drug interaction between imipenem and cilastatin
- Chronic exposure to excess iron promotes EMT and cancer via p53 loss in pancreatic cancer
- Research and development of drug delivery systems based on drug transporter and nano-formulation
- Amino acid transporters:Emerging roles in drug delivery for tumor-targeting therapy