
Stimulatory effect on the transport mediated by organic anion transporting polypeptide 2B1
2020-05-15 09:35
Tohoku University Hospital, Department of Pharmaceutical Sciences, Miyagi 980-8574, Japan
Keywords: OATP2B1 Drug interaction Stimulatory effect Membrane translocation Conformational change
ABSTRACT Drug-drug interaction (DDI) is one of causes of adverse drug events and can result in lifethreatening consequences.Organic anion-transporting polypeptide (OATP) 2B1 is a major uptake transporter in the intestine and contributes to transport various clinically used therapeutic agents.The intestine has a high risk of DDI,because it has a special propensity to be exposed to a high concentration of drugs.Thus,understanding drug interaction mediated by OATP2B1 in the absorption process is important for the prevention of adverse drug events,including decrease in the therapeutic effect of co-administered drugs.Acute drug interaction occurs through the direct inhibitory effect on transporters,including OATP2B1.Moreover,some compounds such as clinically used drugs and food components have an acute stimulatory effect on transport of co-administered drugs by OATP2B1.This review summarizes the acute stimulatory effect on the transport mediated by OATP2B1 and discusses the mechanisms of the acute stimulatory effects of compounds.There are two types of acute stimulatory effects,substrate-independent and -dependent interactions on OATP2B1 function.The facilitating translocation of OATP2B1 to the plasma membrane is one of causes for the substrate-independent acute stimulatory effect.On the contrary,the substrate-dependent effect is based on the direct binding to the substrate-binding site or allosteric progesterone-binding site of OATP2B1.
1.Introduction
Polypharmacy,frequently defined as the use of five or more medicines,is becoming a concerning issue worldwide.With the increase in patients with more complex and multiple diseases,many drugs are prescribed by many different specialists or are prescribed on the basis of the evidence related to each single disease.Inappropriate polypharmacy is a particular concern in elderly population aged above 65 years,because it tends to be associated with adverse drug events.Adverse drug events have been recently estimated to cause approximately 8.7%–16.6% of hospital admissions in a population of elderly [1,2].Drug-drug interaction (DDI) is defined by the Food and Drug Administration (FDA) as follows:“DDI can lead to changed systemic exposure,resulting in variations in drug response of the co-administered drugs.In addition to co-administration of other drugs,concomitant ingestion of dietary supplements or citrus fruit or fruit juice could also alter systemic exposure of drugs,thus leading to adverse drug reactions or loss of efficacy”[3].DDI can result in life-threatening conditions,including bleeding,hypotension,central nervous system depression,considerable electrolyte disturbance,and hypoglycemia [4].DDIs are related to the number of medicines used for treatment.As the number of medicines increases,the potential risk for DDIs increases.For example,the probability of at least one CYP-mediated DDI was 50% for persons taking 5–9 drugs,81% for persons taking 10–14 drugs,92% for persons taking 15–19 drugs,and 100% for persons taking 20 or more drugs [5].The prevalence of polypharmacy from 1997 to 2012 increased from 17.8%to 60.4% in people aged ≥65 years in Ireland [6].This trend is similar in other countries [7,8].Recently,the trend of polypharmacy from 2010 to 2016 in Japan has been reported[9].The total rates of polypharmacy peaked in 2012–2013.The prevalence of polypharmacy in Japan is 16.4%–17.9% in people aged 65–79 years and 28.6%–33.0% in those aged ≥80 years.
The intestine has a high risk of DDIs,because it has a special propensity to be exposed to high concentrations of drugs.Thus,changes in the functions of drug transporters substantially influence the absorption of administered drugs from the intestine.Organic anion transporting polypeptide (OATP) 2B1 has historically been assumed as a major transporter contributing to absorption process in the intestine [10],but apical/basolateral localization and direction of transport in enterocytes remains controversial[10–13].OATP2B1 transports many clinically used drugs such as atorvastatin [14],fexofenadine [15],fluvastatin[16],glibenclamide [17],rosuvastatin [18],and sulfasalazine[19].However,OATP2B1 shows a large substrate-specific difference in affinity of substrate.This may be,at least in part,explained by the observation that OATP2B1 likely has multiple binding sites [20–22].The uptake of estrone-3-sulfate(E3S) into OATP2B1-transfected oocytes showed high and low affinity-binding sites.OATP2B1 shows sodium-independent and pH-dependent transport,though the effect of pH on OATP2B1-mediated transport is substrate-dependent [10,11].The OATP2B1-mediated uptake of E3S and statins appeared to be dependent on pH,but the uptake of bromosulfophthalein(BSP) appeared to be independent on pH [16,23].This may be also explained by the presence of multiple binding sites,because only the low affinity site shows the pH sensitive transport of substrates [20].
In 2002,Dresser et al.reported that grapefruit,orange,and apple juice caused inhibition of absorption of fexofenadine in healthy volunteers,though they postulated that the mechanism of action was the inhibition of OATP1A2 by the fruit juices [24].Examples of clinical relevance of DDI by OATP2B1 are gradually increasing [25,26].A number of pharmacogenetics (PGx) studies have investigated the impact of OATP2B1 polymorphisms on the pharmacokinetics (PK)and pharmacodynamics (PD) of drugs [27–33].The details of clinical relevance of OATP2B1 are well documented by Yu et al.[34]and Wu et al.[26].Thus,understanding drug interaction mediated by OATP2B1 in the absorption process is important for prevention of adverse drug events,including decrease in the therapeutic effect of co-administered drugs [34,35].
In most cases,acute DDI occurs through the direct inhibitory effect (Fig.1 A) or by allosteric binding (Fig.1 B)on transporters,including OATP2B1 [36–38].Components of apple,orange,and grapefruit juice such as naringenin can directly inhibit OATP2B1 [36].Those DDIs can occur through simultaneous oral administration.Moreover,there are other types of DDIs involving changes in the expression level of transporter [39](Fig.1 C) and in the localization of transporter[40](Fig.1 D) and in the degradation of transporter [41](Fig.1 E).The DDIs caused by an expression change and protein degradation (Fig.1 C and 1E) occur with one to several hours after the treatment of drug to the cells.Thus,those DDIs may not occur in the intestine through simultaneous oral administration.However,the DDI caused by a localization change (Fig.1 D) is an acute effect.It occurs within 1–60 min after the dosage of drug,indicating that the type of DDIs may occur in the intestine by simultaneous oral administration.In this review,we use the expression“acute DDI”when the changes in transport or absorption occur through simultaneous treatment (invitro) or administration(invivo).We,therefore,focus on the newly found DDI acute stimulatory effect on OATP2B1.
2.Acute stimulatory effect on the uptake of OATP2B1 substrates
Pizzagalli et al.reported the acute stimulatory effects of prostaglandin (PG) A1 and PGA2 on OATP2B1 function [42].Their study was the first report about an acute stimulatory effect on OATP2B1.The stimulatory effect of PGA1 was detectable for 15 s,and no further stimulation occurred after treatment for longer time.PGA2 also had a stimulatory effect on OATP2B1 but PGE1,PGE2,and PGJ2 did not.Cyclopentenone,which is a ring structure contained into PGA1 and PGA2,(Fig.2) showed a stimulatory effect on the transport mediated by OATP2B1.However,the cyclopentenone ring did not fully contribute to stimulate the transport mediated by OATP2B1,because PGJ2,which contains a cyclopentenone ring (Fig.2),did not show any stimulatory effect.Although the mechanisms of the stimulatory effect of PGA1 and PGA2 were not clarified,PGA1 and PGA2 did not change the affinities (K m s) of E3S and dehydroepiandrosterone sulfate(DHEAS) but did change the V max of those substrates.These findings suggested that PGA1 and PGA2 might not induce the conformational change in OATP2B1.Several studies also reported acute stimulatory effects of some drugs or food components on OATP2B1,though they did not investigate the mechanism in detail (Table 1).It is important to consider that the experimental concentration of the listed compounds reflected in clinical settings.Table 2 shows the clinical concentrations of the endogenous stimulators for OATP2B1 function.The concentrations of all those endogenous stimulators are 100–1 000 000-fold different between the experimental and clinical concentrations.Thus,the possibilities of the stimulation of OATP2B1 function by those endogenous stimulators are low.On the contrary,orally administered drugs and food components should be considered at their intestinal concentration.Thus,the expected intestinal concentrations,which are calculated according to the FDA guidance [58]or European Medicines Agency (EMA) guideline [59],are listed in Table 3 .Moreover,Ichijo et al.recently reported that orally administered water reached a steady state within 30 min in the jejunum and ileum [60].Therefore,the expected intestinal concentrations,which are calculated based on the small intestinal water content (206 g) in humans [61],are also listed in Table 3 .The concentrations of all stimulators reached experimental concentrationsinvitroexperiments,indicating that all the listed stimulators may be able to facilitate the absorption of OATP2B1 substrates in clinical setting.However,it is difficult to confirm the molecular mechanisms of DDI in clinical settings,because phenomena similar as the stimulation of OATP2B1 function in the intestine also occur through the inhibition of metabolic enzymes such as CYP3A4,the inhibition of efflux pumps such as P-gp,MRPs,and BCRP in the intestine,the stimulation of other uptake transporters such as ASBT in the intestine,and the inhibition of OATPs such as OATP1B1 and OATP1B3 in the liver.All those situations induce the increase in plasma concentration of the drug.On the other hand,invitroexperiments are better methods for the clarifying the molecular mechanisms of the stimulation of the uptake to the cells.Therefore,we selected some studies that investigated the stimulatory effect on OATP2B1 in detail.
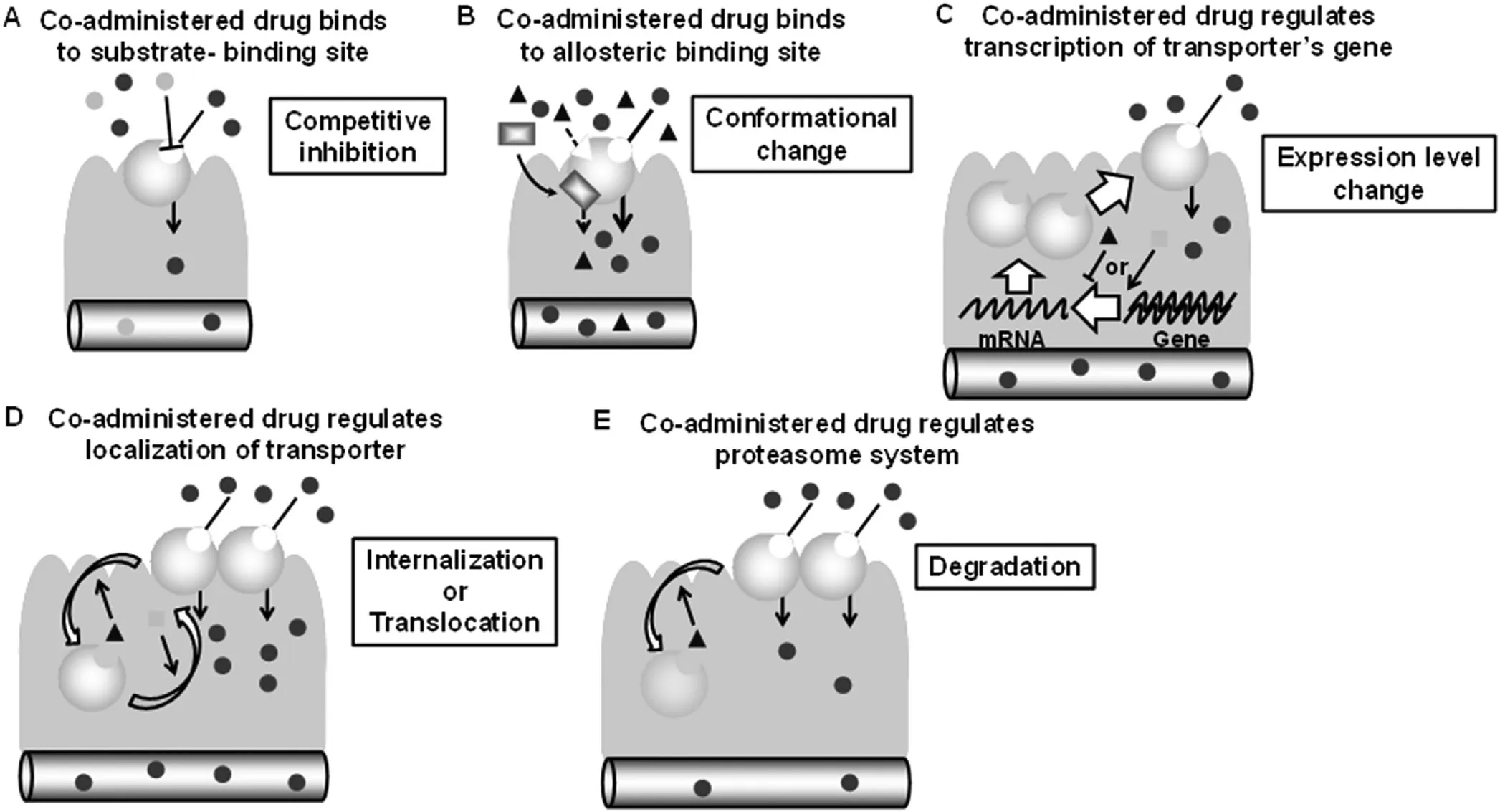
Fig.1–Schematic diagrams for various types of DDI in the intestinal transporter.
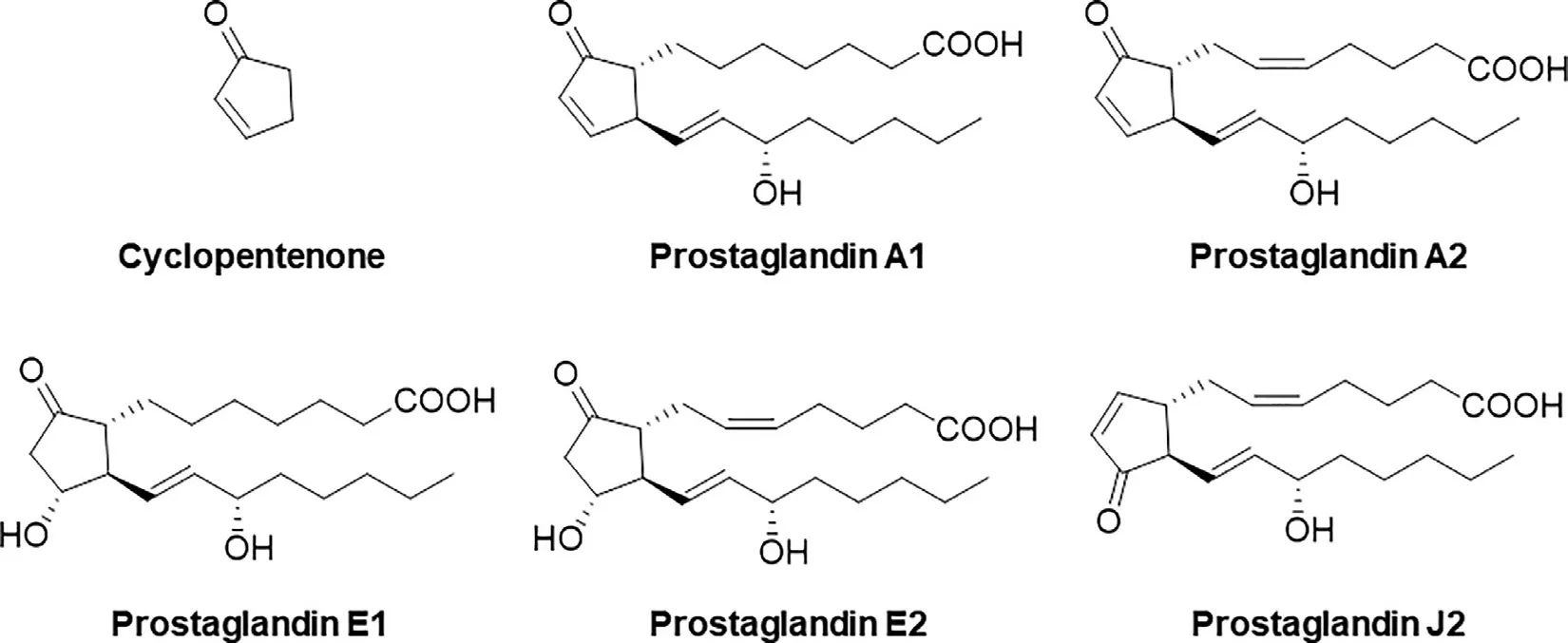
Fig.2–Chemical structures for Prostaglandins.

Table 1–Facilitating effect of drugs or food components on the uptake of OATP2B1 substrates.
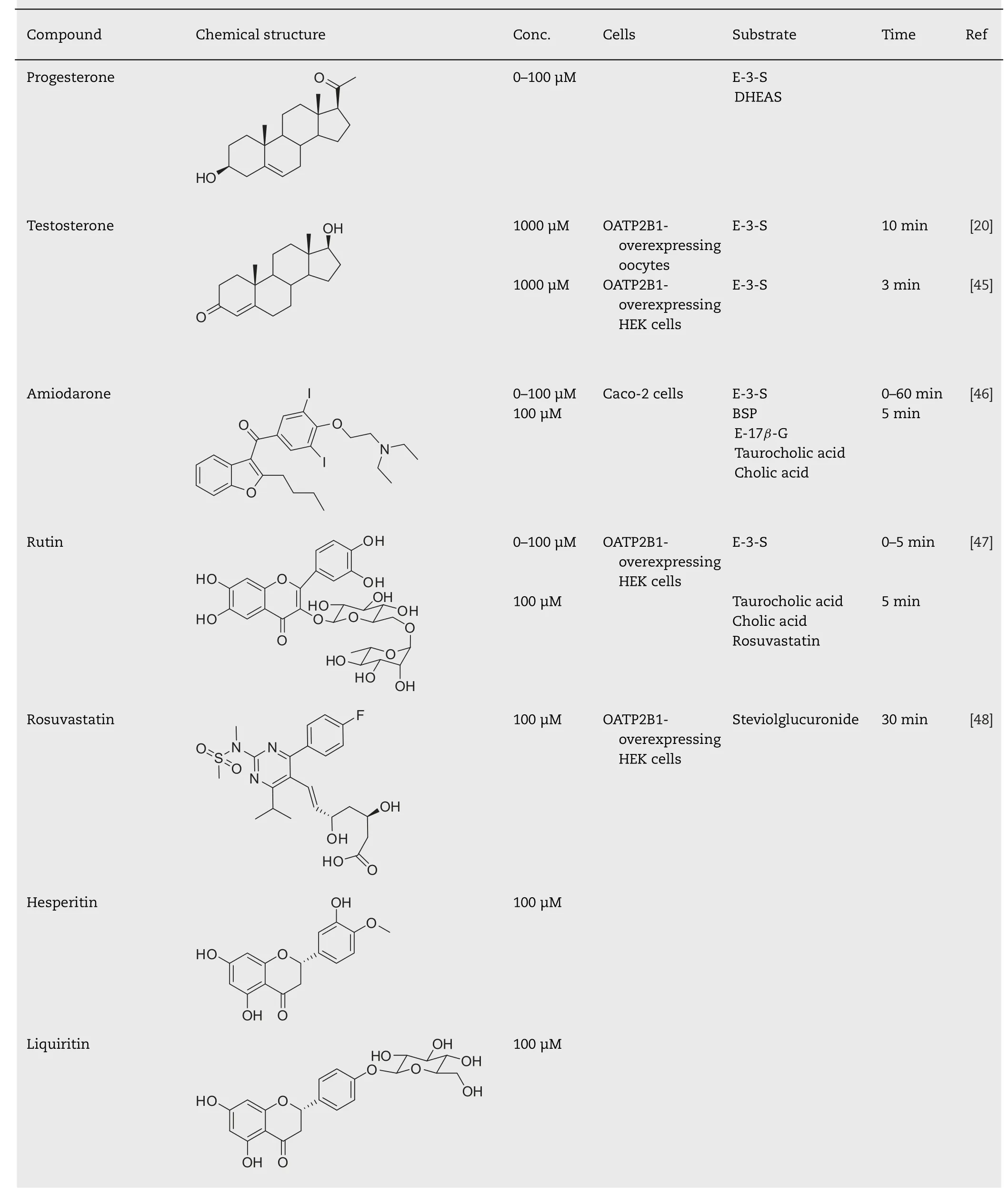
Table 1 (continued)
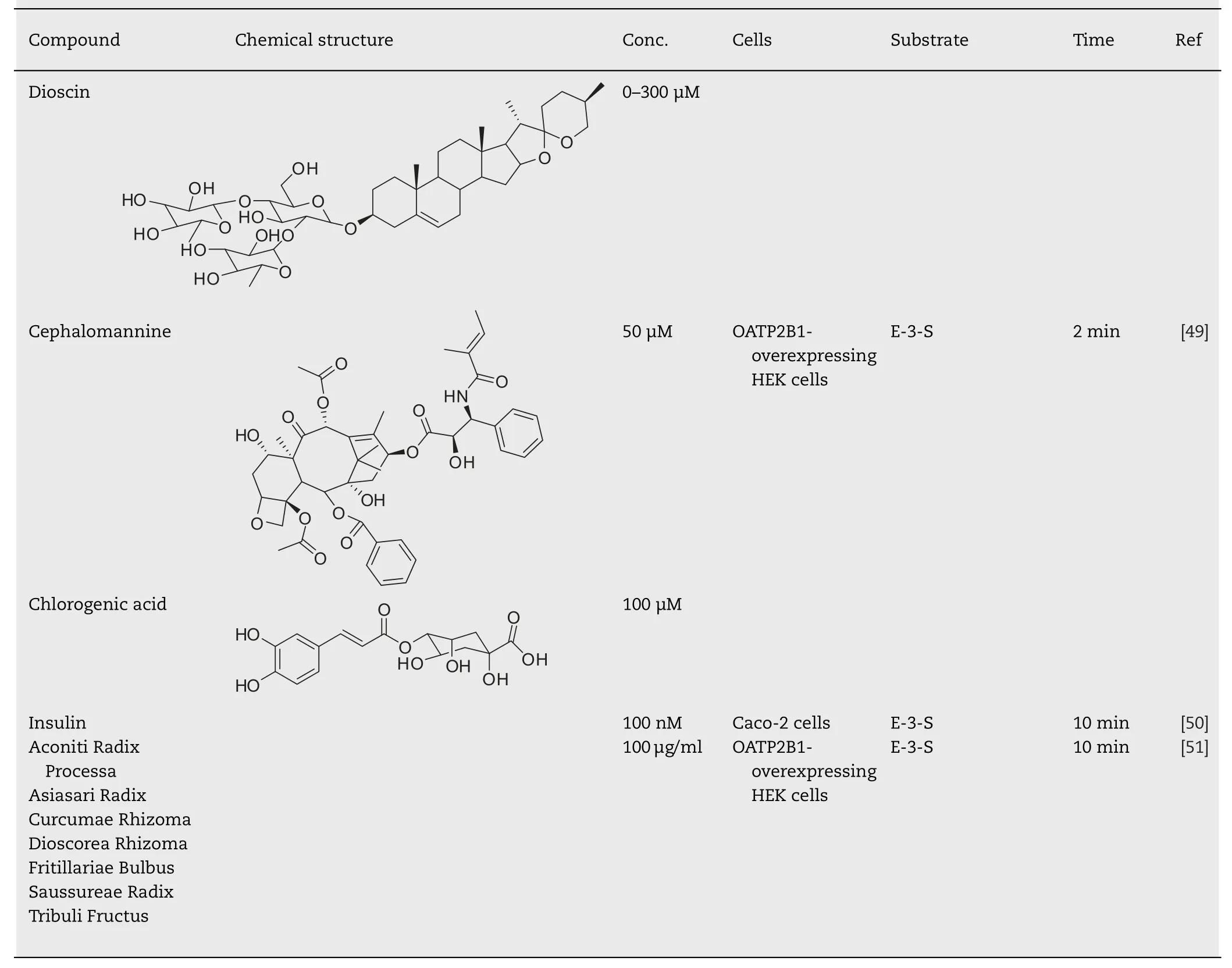
Table 1 (continued)
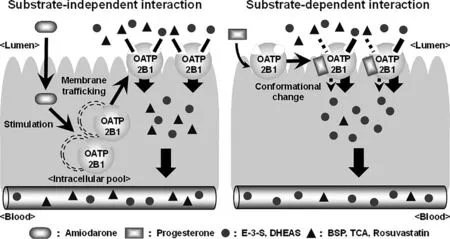
Fig.3–Schematic diagrams for substrate-independent and -dependent effect on OATP2B1 function.
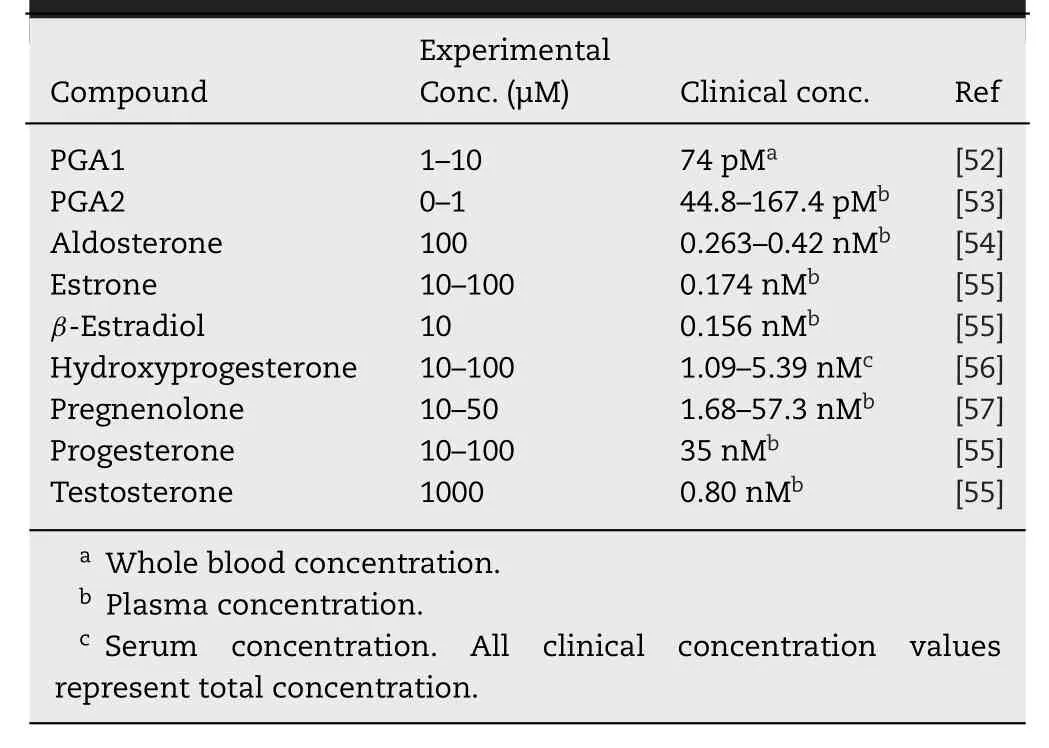
Table 2–Clinical concentration of the endogenous stimulator for the transport mediated by OATP2B1.
3.Stimulatory effect on the uptake of OATP2B1 substrates in substrate-independent manner
Amiodarone (AMD),which is a potent drug used in the treatment of serious supraventricular and ventricular tachyarrhythmias,is known to be a substrate for OATP2B1[67],indicating that AMD can bind to the substrate-binding domain of OATP2B1.However,AMD surprisingly increased the uptake of OATP2B1 substrates by the simultaneous treatment in Caco-2 cells [46].AMD did not change the affinity (Km)of estrone sulfate,but it did change its V max as well as PGA1 or PGA2 [42,46].This acute stimulatory effect of AMD on OATP2B1 substrates was caused by the stimulation of the translocation of OATP2B1 to the plasma membrane.This finding is consistent with the kinetic analysis and indicates the acute stimulatory effect of AMD on OATP2B1 as a substrate-independent effect.Indeed,AMD can stimulate the transport of other OATP2B1 substrates such as E3S,BSP,estradiol-17β-glucuronide (E-17β-G),taurocholic acid(TCA),and cholic acid (CA) [46].The stimulatory effect of AMD occurred strongly and rapidly in aninvivoexperiment.When AMD is co-administered with BSP to male Wistar rats,absorption of BSP is significantly increased,and AUC 0–105 min and C max after intraintestinal administration are 3.7-fold and 3.4-fold higher than those in control rats,respectively.It is difficult to confirm the target of the stimulatory effect of AMDinvivoexperiments,but some evidences indicate that the stimulatory effect of AMD targets OATP2B1.BSP is not metabolized by CYP enzymes,suggesting that the stimulatory effect of AMD does not target CYP enzymes.AMD does not affect the basolateral to apical transport of E-3-S in Caco-2 cells.In addition,inhibitors for ABC transporters do not affect the stimulatory effect of AMDinvitroexperiments,suggesting that the stimulatory effect of AMD does not target ABC transporters.AMD facilitates the initial absorption rate of BSP and does not affect the pharmacokinetics of BSP after intravenous administration,suggesting that the stimulatory effect of AMD does not target hepatic transporters.OATP1A2 mRNA and protein expression in the intestine are very low compared to those of OATP2B1 [68,69],suggesting that the contribution of OATP1A2 to the stimulatory effect of AMD may be subtle.All these findings indicate that the increased OATP2B1 expression at the apical membrane is a dominant factor for facilitating the absorption of BSP by co-administered AMD.
The acute stimulating effect on the translocation of OATP2B1 to the plasma membrane is not AMD-specific.Rutin,a flavonol glycoside compound contained in some foods such as tomatoes,red wine,and,at high concentrations,in buckwheat also has a stimulatory effect on the translocation of OATP2B1 to the plasma membrane [47]Thus,rutin can acutely facilitate the uptake of OATP2B1 substrates in a substrate-independent manner in HEK293 cells overexpressing OATP2B1.However,the treatment with rutin did not affect the uptake of E-3-S in Caco-2 cells,used as a model of intestinal epithelium.This may reflect the lower bioavailability of rutin compared to that of other quercetin glucosides such as quercetin 3-O-β-glucoside (isoquercitrin)and quercetin 4′-O-β-glucoside (spiraeoside) [70,71].
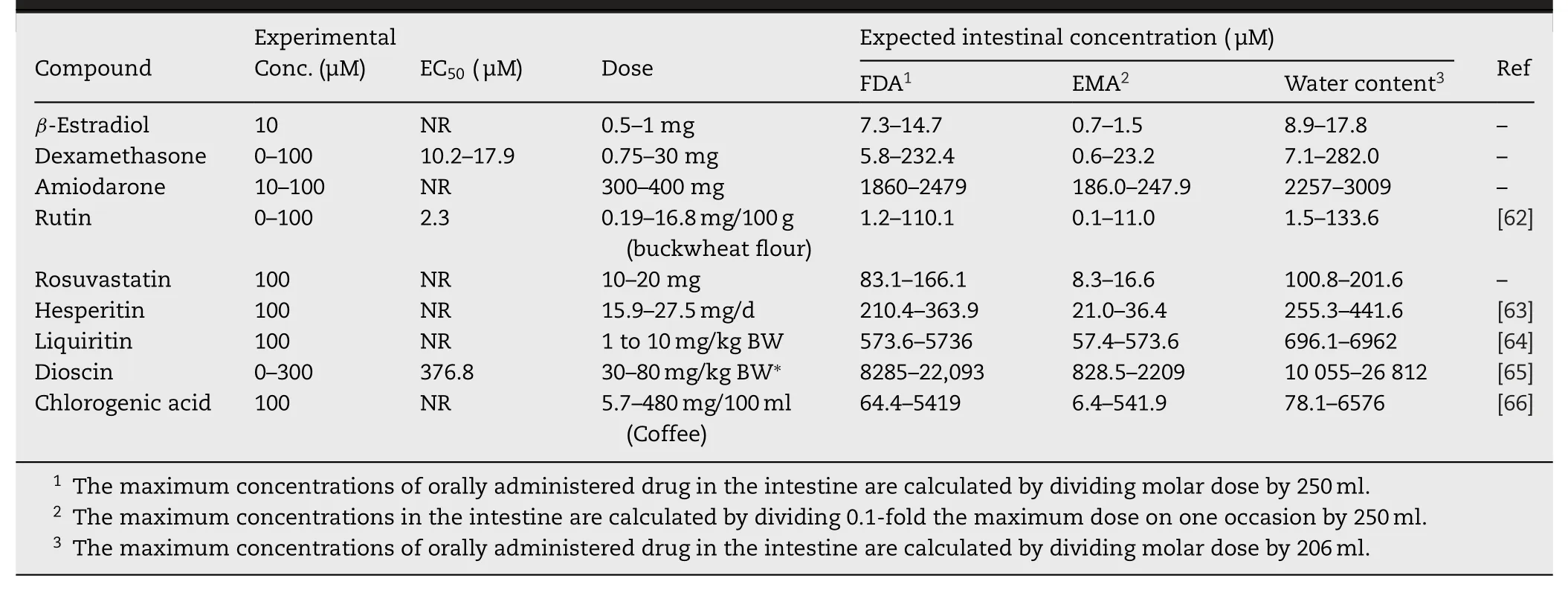
Table 3–Expected intestinal concentration of the exogenous stimulator for the transport mediated by OATP2B1.
4.The mechanism of the translocation of OATP2B1 to the plasma membrane
The small GTPase Rab protein family is known to be involved in membrane trafficking of proteins [72,73],including transporters [74–79].Among those family members,Rab8a is highly expressed in the small intestine and contributes to the localization of several transporters such as sodiumdependent vitamin C transporter 1 (SVCT1),sodiumdependent glucose transporter 1 (SGLT1),and peptide transporter 1 (PEPT1),to the apical membrane [80–83].OATP2B1 is also regulated by Rab8a [50].The uptake of E-3-S mediated by OATP2B1 increased in Rab8a-overexpressed cells and decreased in Rab8a knocked down cells.These findings indicate that Rab8a is one of the important factors for the OATP2B1 translocation to the plasma membrane.Rab8a is well known to be activated by insulin signaling,resulting to stimulate the translocation of glucose transporter(GLUT) 4 to the plasma membrane and facilitate the uptake of glucose [84–88].The mechanism is one of the most important mechanisms for reducing effect of insulin on the blood glucose.Insulin activates Rab8a within 2 min[87]and stimulates the translocation of GLUT4 to the plasma membrane for 10–30 min.In the same manner,insulin facilitated the uptake of E-3-S by the treatment for just 10 min [50].At the same time,the expression level of OATP2B1 in the plasma membrane increased.Rab8a shows the highest expression in the intestine (The data from The Human Protein Atlas;https://www.proteinatlas.org).AMD stimulated the absorption of BSP after intraintestinal administration but did not affect the pharmacokinetics of BSP after intravenous administration.These findings indicate that AMD has no effect on hepatic OATP2B1invivoexperiments,though OATP2B1 is expressed in both liver and intestine.The distribution of Rab8a in the body may cause the differences in the effect of AMD in the intestine and liver.
5.Stimulatory effect on the uptake of OATP2B1 substrates in substrate-dependent manner
OATP2B1 is known as transporter for sulfated steroids such as E-3-S,DHEAS,and pregnenolone sulfate (PS).Grube et al.reported that the transport of sulfated steroids by OATP2B1 was enhanced by the treatment of the steroid hormone progesterone for just 5 min [43].In contrast to the stimulatory effects of AMD or rutin,progesterone could stimulate the uptake of sulfated steroids by OATP2B1 but not the uptake of BSP,atorvastatin,and glibenclamide [44].These findings suggest that the mechanism of stimulatory effect of progesterone is different from that of AMD or rutin.Because the K m value changed by the presence of progesterone [43],the stimulatory effect of progesterone might be based on a direct binding to the substrate-binding site of OATP2B1 or an allosteric progesterone-binding site,like that in P-gp[89],and,then,a change in the conformation.OATP2B1 has multiple binding sites for substrates within the molecule [20–22].Although progesterone is not a substrate for OATP2B1 [43],it is a possible that progesterone binds a substrate-binding site and changes the conformation of the other binding site.Indeed,progesterone stimulated only the uptake of E-3-S by the high-affinity site but inhibited that by the low-affinity sites [22].These findings do not exclude the possibility that progesterone binds an allosteric binding site,though allosteric binding sites in OATP2B1 have not been identified.Further studies are needed to reveal the mechanism of the stimulatory effect of progesterone on the transport by OATP2B1.Moreover,the relevance of the stimulatory effect of progesterone in a living body remains unknown.
6.Conclusion
Although the information on the acute stimulatory effect on transporter function is scarce compared to that on direct transporter inhibition,the mechanism of the acute stimulatory effect on transporter function is been gradually elucidated.There are two types of acute stimulatory effects,substrate-independent and -dependent interactions on OATP2B1 function (Fig.3).The facilitating translocation of OATP2B1 to the plasma membrane is one of causes for the substrate-independent acute stimulatory effect.On the contrary,the substrate-dependent effect is based on the direct binding to the substrate-binding site or allosteric progesterone-binding site of OATP2B1.However,we do not have any information of clinical relevance for the acute stimulatory effects of drugs or food components.Only AMD facilitates the absorption OATP2B1 substrateinvivoexperiments.Further studies are needed to reveal the clinical importance of the stimulatory effect of drugs or food components on OATP2B1 in a living body.However,there are some problems such as a large substratespecific difference in potency and an overlapping in the substrate specificities to clarify the clinical relevance of DDI mediated by OATP2B1,as point out by Wu et al.[26].Briefly,because of the significant overlap in the substrate specificities of OATP2B1 and other OATPs,no selective substrate or inhibitor has been identified.Although the differences in the affinity of drugs for OATP2B1 and other OATPs are useful to solve the problem,development of a selective substrate/inhibitor for OATP2B1 is necessary in the future.
Conflicts of interest
The authors report no conflicts of interest.The authors alone are responsible for the content and writing of this article.
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.This review was in part supported by the Japan Society for the Promotion of Science (JSPS) Kakenhi grant number 19K21219 .
 Asian Journal of Pharmacentical Sciences2020年2期
Asian Journal of Pharmacentical Sciences2020年2期
- Asian Journal of Pharmacentical Sciences的其它文章
- The solute carrier transporters and the brain:Physiological and pharmacological implications
- The role of transporters in cancer redox homeostasis and cross-talk with nanomedicines
- Intestinal OCTN2-and MCT1-targeted drug delivery to improve oral bioavailability
- Pharmacologic inducers of the uric acid exporter ABCG2 as potential drugs for treatment of gouty arthritis
- Amino acid transporters:Emerging roles in drug delivery for tumor-targeting therapy
- Glutamine transporters as pharmacological targets:From function to drug design