
Pharmacologic inducers of the uric acid exporter ABCG2 as potential drugs for treatment of gouty arthritis
2020-05-15 09:35
Department of Cell Biology and Biochemistry, Texas Tech University Health Sciences Center, Lubbock, TX 79430, United States
Keywords: Uric acid excretion Intestine ABCG2 Loss-of-function mutations Gouty arthritis Pharmacologic inducers
ABSTRACT Uric acid is the end product of purine catabolism and its plasma levels are maintained below its maximum solubility in water (6–7 mg/dl).The plasma levels are tightly regulated as the balance between the rate of production and the rate of excretion,the latter occurring in urine (kidney),bile (liver) and feces (intestinal tract).Reabsorption in kidney is also an important component of this process.Both excretion and reabsorption are mediated by specific transporters.Disruption of the balance between production and excretion leads to hyperuricemia,which increases the risk of uric acid crystallization as monosodium urate with subsequent deposition of the crystals in joints causing gouty arthritis.Loss-of-function mutations in the transporters that mediate uric acid excretion are associated with gout.The ATP-Binding Cassette exporter ABCG2 is important in uric acid excretion at all three sites:kidney (urine),liver (bile),and intestine (feces).Mutations in this transporter cause gout and these mutations occur at significant prevalence in general population.However,mutations that are most prevalent result only in partial loss of transport function.Therefore,if the expression of these partially defective transporters could be induced,the increased number of the transporter molecules would compensate for the mutation-associated decrease in transport function and hence increase uric acid excretion.As such,pharmacologic agents with ability to induce the expression of ABCG2 represent potentially a novel class of drugs for treatment of gouty arthritis.
1.Introduction
Uric acid is endogenously as the end product of purine catabolism.When the purine free bases guanine and adenine cannot be salvaged to generate purine nucleotides,they are degraded by xanthine oxidase to generate uric acid.At physiological levels and under hydrophilic conditions,uric acid functions as a potent antioxidant.In fact,it is the primary antioxidant in circulation in humans.Of relevance to this issue is the fact that xanthine oxidase function that produces uric acid is also associated with generation of reactive oxygen species superoxide while converting hypoxanthine to xanthine,and hydrogen peroxide while converting xanthine to uric acid.It is of significant biological curiosity that the same enzymatic step is involved in the production of oxidant molecules as well as a potent antioxidant.Uric acid is also an iron chelator;this also contributes to the antioxidant function of uric acid because free iron in its divalent form (Fe2+) is an oxidant due to its ability to participate in Fenton reaction(Fe2++H2O2→ Fe3++OH•+OH−),which generates the highly reactive hydroxyl radical.Paradoxically,uric acid is also a prooxidant when present in an hydrophobic environment;in this capacity,uric acid can cause oxidative stress,endothelial dysfunction,and inflammation.
The plasma levels of uric acid are tightly controlled and maintained within the range of 3.5–7.0 mg/dl (200–400 μM)in men and 2.5–6.0 mg/dl (150–350 μM) in women [1,2].This physiological range is important because the maximum solubility of uric acid in aqueous media is 6–7 mg/dl [2].When the concentration in circulation goes beyond this maximal solubility value,uric acid crystallizes as monosodium urate,which gets deposited in joints and cause inflammation and gouty arthritis [3,4].These crystals are phagocytosed into monocytes/neutrophils to initiate inflammation and arthritic pathology.Therefore,the plasma levels of uric acid are tightly controlled as the balance between the rate of production and the rate of excretion.Xanthine oxidase,the enzyme that generates uric acid,is expressed predominantly in the liver and intestine in humans and other mammals [5].Liver is the primary site for catabolism of endogenous free purine bases whereas small intestine is responsible for catabolism of excess diet-derived purine bases.Excretion of uric acid occurs mostly at three sites:kidney (urine),intestine (feces),and liver (bile).A major fraction (~90%) of uric acid filtered at the glomerulus is reabsorbed,which is a critical determinant of plasma levels of uric acid in addition to the rate of production and the rate of excretion.When the rate of production(and reabsorption) exceeds the rate of excretion,uric acid accumulates,causing hyperuricemia and increasing the risk of uric acid crystallization and consequent gouty arthritis.This could be due to increased production (and reabsorption),decreased excretion or both.Gout represents the most common inflammatory arthritis;it affects millions of people in the United States.Hyperuricemia is also associated with metabolic syndrome,diabetes,and cardiovascular disease.
2.Transporters involved in the handling of uric acid in vivo
Uric acid possesses two ionizable groups with pKa values of 5.4 and 10.3;as such,this metabolite exists as a monovalent urate anion under physiological conditions with pH 7.4[6,7].This ionized form is not diffusible across biological membranes;therefore,both excretion and reabsorption processes involve specific transporters [8–14].Kidney,which participates both in reabsorption as well as excretion of uric acid,expresses all of these transporters (Table 1,Fig.1).In contrast,liver and intestine,which participate preferentially in uric acid excretion,express only a few of these transporters:liver–MRP4 (sinusoidal membrane) and ABCG2 (canalicular membrane);intestine–MRP4 (apical membrane and basolateral membrane),SLC2A9 (basolateral membrane and apical membrane),and ABCG2 (apical membrane) (Fig.2).
3.Reabsorption in kidney
In the renal tubule,reabsorption of uric acid from the glomerular filtrate consists of two steps:uptake across the brush-border membrane via the anion transporters SLC22A12(also known as urate anion transporter 1 or URAT1) and SLC22A11 (also known as organic anion transporter 4 or OAT4) and release from the cells into systemic circulation across the basolateral membrane via the voltage-sensitive,high-capacity,anion transporter SLC2A9 (also known as GLUT9) (Fig.1) [15,16].GLUT9 has two splice variants,namely GLUT9a and GLUT9b.Both variants are expressed in renal tubular cells:GLUT9a in the basolateral membrane and GLUT9b in the apical membrane.As GLUT9 is an electrogenic urate transporter,both variants mediate the transport of urate out of the cells because of the insidenegative membrane potential that promotes the function of any transport process involved in anion removal from cells.Thus,GLUT9a in the basolateral membrane functions in the renal reabsorption of urate.URAT1 is an anion exchanger that mediates the influx of urate into cells coupled to the efflux of monovalent organic anions out of the cells;the physiologically relevant anion in this exchange with urate is lactate.As a consequence,the function of URAT1 in kidney is coupled to the functions of SLC5A8 (also known as sodium-coupled monocarboxylate transporter 1 or SMCT1)and SLC5A12 (SMCT2),both mediating Na+-coupled active entry of lactate from glomerular filtrate into renal tubular cells [17–19],thus providing lactate for subsequent exchange with urate via URAT1.This functional coupling is supported by the findings that loss of function of both Slc5a8 and Slc5a12 in kidney decreases the ability of URAT1 to absorb urate,thus resulting in increased excretion of uric acid in urine [20].A similar phenomenon occurs with OAT4,which is also an anion exchanger but with preference for divalent organic anions such asα-ketoglutarate and succinate.The basolateral membrane of the tubular cells express a highaffinity concentrative uptake system for these dicarboxylates,known as NaDC3 (Na+/dicarboxylate cotransporter 3 or SLC13A3) [21,22],which accumulates dicarboxylates in cells by uptake from blood and these divalent anions serve as the exchangeable substrates for OAT4 in the brush-border membrane.
4.Excretion in kidney
Excretion of uric acid in kidney also consists of two steps:entry from circulation into tubular cells across the basolateral membrane via the anion transporters SLC22A6 (also known as organic anion transporter 1 or OAT1) and SLC22A8 (OAT3) and exit from the cells into tubular lumen across the brush-border membrane via GLUT9b,phosphate transporters SLC17A1 (also known as Na+-dependent phosphate transporter 1 or NPT1)and SLC17A3 (NPT4) as well as the ATP Binding Cassetteexporters ABCG2 (also known as breast cancer resistance protein or BCRP) and ABCC4 (also known as multidrug resistance related protein 4 or MRP4) (Fig.1) [8–13].OAT1 and OAT3 are anion exchangers with preference for divalent organic anions such asα-ketoglutarate and succinate as the exchangeable substrates to promote urate entry from blood into cells;Thus,the function of these transporters is coupled to the function of NaDC3.NPT1 and NPT4 were originally identified as Na+-dependent phosphate transporters,but now are considered to be Na+-independent electrogenic anion transporters;they transport urate out of the cells into tubular lumen driven by the inside-negative membrane potential that exists across the brush-border membrane [23,24].MRP4 and ABCG2 are unidirectional exporters of urate out of the cells into tubular lumen,and their transport function is coupled to energy from ATP hydrolysis.
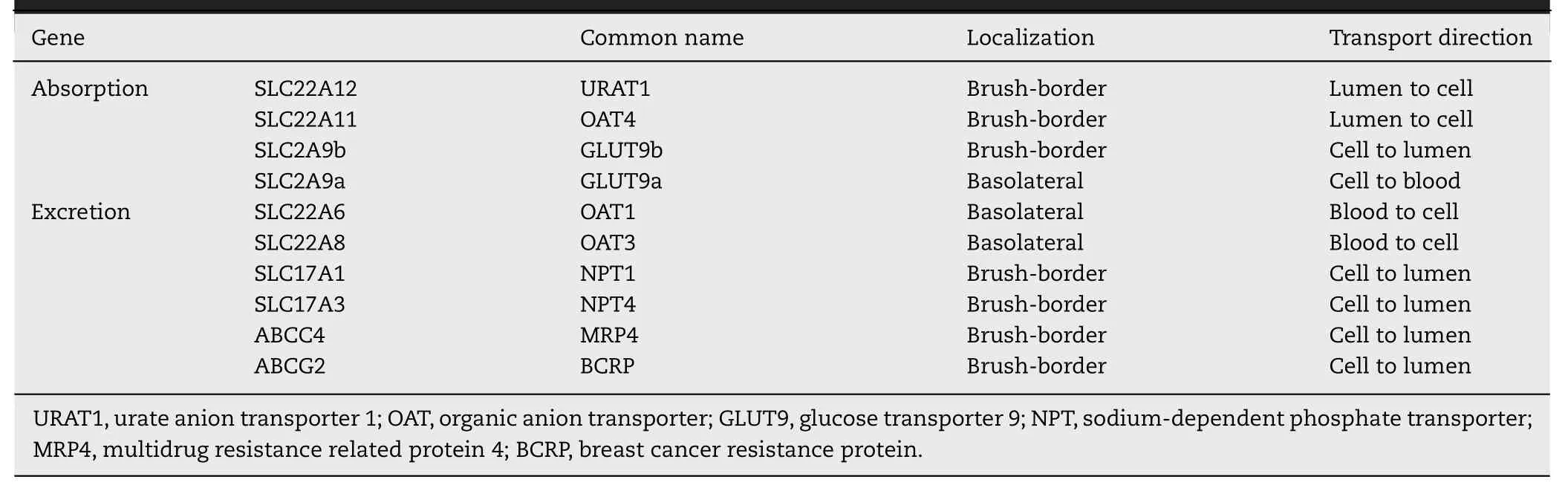
Table 1–Urate transporters in renal tubular epithelium.
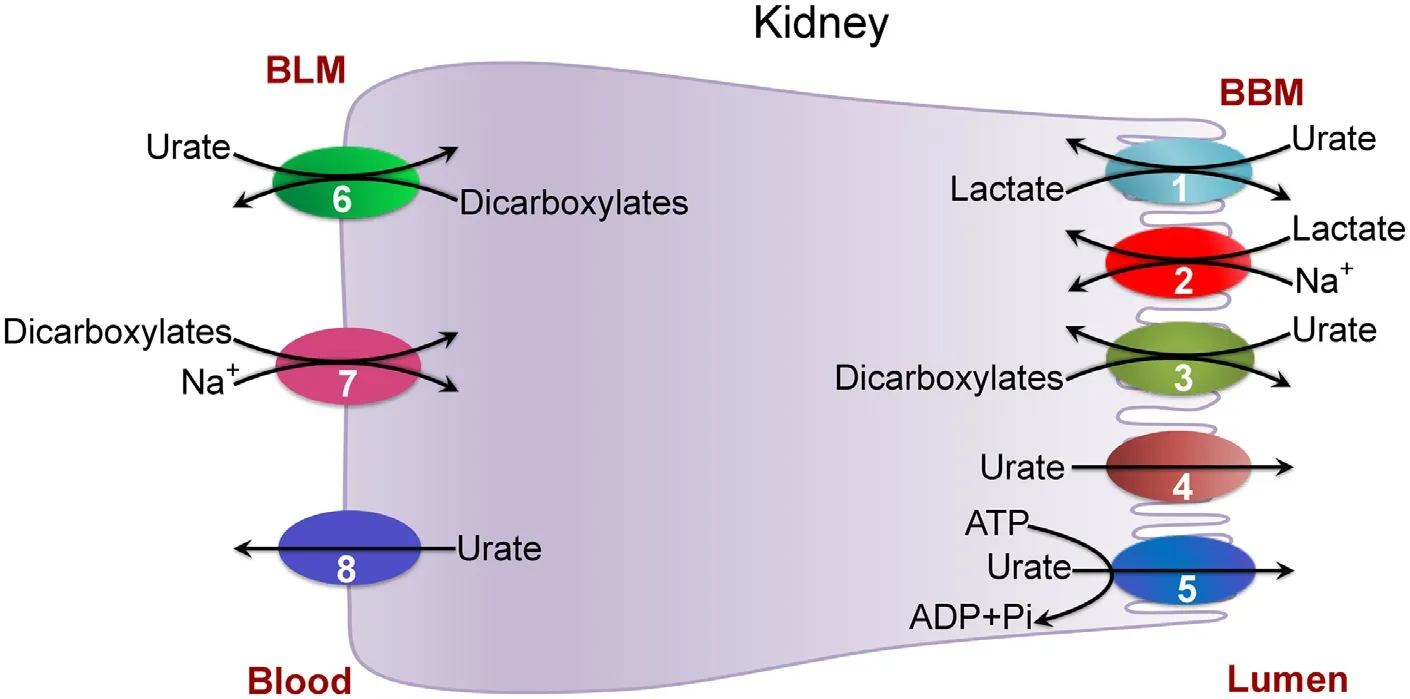
Fig.1–Transporters involved in the handling of urate in kidney.BLM,Basolateral membrane;BBM,Brush-border membrane(apical membrane);1,URAT1;2,SMCT1/SMCT2;3,OAT4;4,NPT1/NPT4/GLUT9;5,ABCG2/MRP4;6,OAT1/OAT3;7,NaDC3;8,GLUT9.
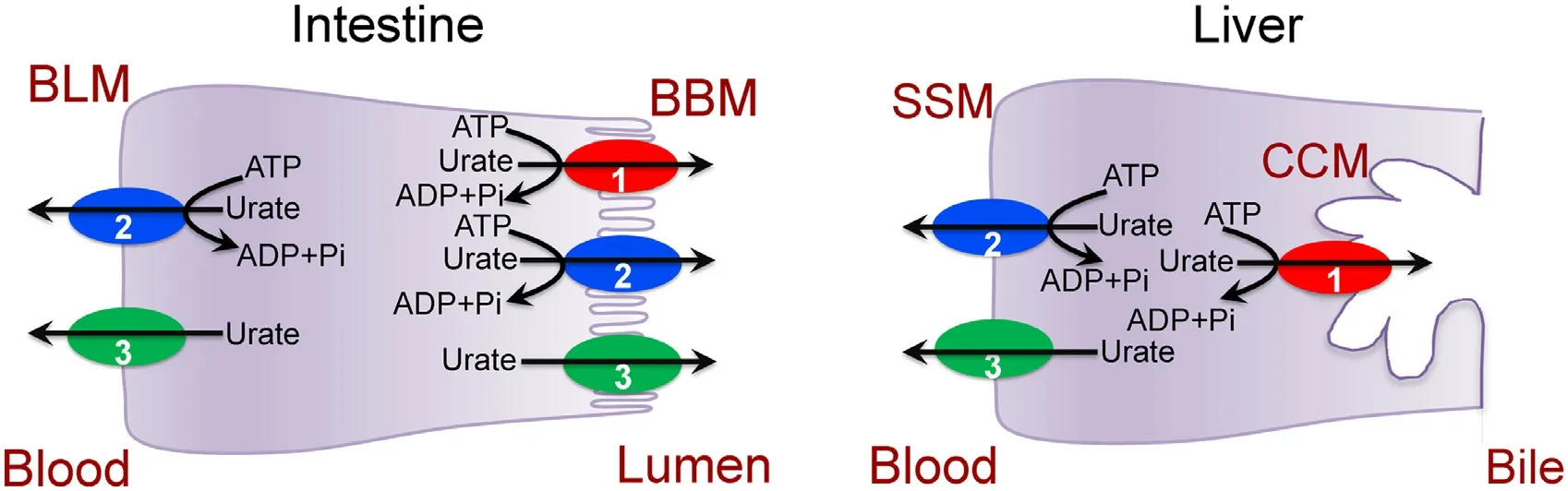
Fig.2–Transporters involved in the handling of urate in intestine and liver.BLM,Basolateral membrane;BBM,Brush-border membrane (apical membrane);SSM,Sinusoidal membrane;CCM,Canalicular membrane.1,ABCG2;2,MRP4;3,GLUT9.
5.Excretion in the intestinal tract
Intestinal tract is not generally regarded as an excretory organ.However,the lumen-facing brush-border membrane of the epithelial cells in the small intestine as well as in the large intestine robustly express the export pump ABCG2 (small intestine>large intestine) whose only known function is to excrete selective endogenous and xenobiotic metabolites from cells into lumen (Fig.2).Most studies on this transporter in the intestinal tract focused on its role in restricting oral bioavailability of pharmacologic agents.However,given the fact that endogenous metabolites such as uric acid,protoporphyrin IX,and heme are substrates for ABCG2,the potential physiological functions of this transporter cannot be ignored.The role of ABCG2 in uric acid excretion is particularly relevant in this regard because the intestinal tract is exposed to large quantities of nucleic acid in the diet as an important source of exogenous purines and also because this tissue expresses high levels of xanthine oxidase which metabolizes most of this diet-derived purines into uric acid.Thus,liver is not the only organ that generates uric acid as generally thought;the intestinal tract represents an equally important site for uric acid production,though from exogenous purines instead of from endogenous purines.It seems that the intestinal tract functions as a barrier against excessive absorption of dietary purines,and it does so by effectively converting these excess purines into uric acid with subsequent elimination into feces.ABCG2 appears to be the major transporter responsible for excretion of uric acid in this tissue [25].Deletion ofAbcg2in mice led to a significant reduction in intestinal elimination of uric acid with concomitant increase in plasma levels of uric acid [26].MRP4,also an ATP-dependent urate exporter,is expressed in the apical membrane as well as in the basolateral membrane of enterocytes.As the small intestine is a major producer of uric acid,MRP4 contributes to the release of urate from the cells into the lumen across the apical membrane as well as into the circulation across the basolateral membrane(Fig.2).The role of GLUT9 in intestinal handling of urate is ambiguous.Mutations in this transporter are associated with reduced levels of uric acid in blood,suggesting that the transporter,expressed in kidney,liver,and intestine,plays a role in retention of uric acid in blood.However,intestinespecific deletion of GLUT9 in mice results in increased uric acid in blood,and these mice show symptoms of early-onset metabolic syndrome [14].It seems that GLUT9,which is expressed both in the apical membrane and the basolateral membrane of polarized renal and intestinal epithelial cells,might be involved in urate reabsorption in kidney but in urate excretion in intestine (Fig.3).
6.Relative contributions of kidney and intestine to whole-body uric acid excretion
Uric acid is eliminated from the body at three sites:kidney,intestine,and liver.Among these three sites,biliary secretion in liver represents the least mode of elimination.Urinary excretion via kidney constitutes 60%–70% of wholebody uric acid elimination,the remainder occurring in the intestinal tract [27,28].ABCG2,being the predominant uric acid exporter expressed throughout the entire intestinal tract,is therefore responsible for 30%–40% of uric acid elimination.The expression of ABCG2 is most robust in jejunum and ileum.
7.Hepatic handling of uric acid
Liver is the primary producer of uric acid from turnover of endogenous purines.The urate exporter MRP4 is expressed in the sinusoidal membrane of hepatocytes (Fig.2),indicating that the release of urate from liver into blood is mediated at least partly by this transporter.The other urate exporter,ABCG2,is expressed only in the canalicular membrane,thus participating in the excretion of urate into bile.GLUT9 is expressed in liver,but its localization and physiological role in the handling of urate remain unexplored.
8.Mutations and variants in genes coding for urate transporters and their relevance to plasma levels of uric acid
Defective function or decreased expression in transporters associated renal reabsorption would decrease the circulating levels of uric acid (hypouricemia) as is the case with loss-of-function mutations in URAT1 [29].Single nucleotide polymorphisms (SNPs) in non-coding regions ofGLUT9are also associated with decreased plasma levels of uric acid and protection against gout [30].It is likely that these polymorphisms impact the stability of mRNA,thus reducing the expression of the transporter protein,an idea that would logically explain the hypouricemic effect of such SNPs,but the validity of this idea has not yet been tested.In contrast to these transporters involved in uric acid reabsorption,defective function in transporters associated with excretion would increase circulating levels of uric acid (hyperuricemia or gout).Polymorphisms in the genes coding for SLC17A1 and SLC17A3 are associated with gout [8–13].Similarly,loss-offunction mutations in ABCG2 also cause gout [26,31–34].
9.ABCG2 as a therapeutic target for a novel class of drugs to treat gouty arthritis
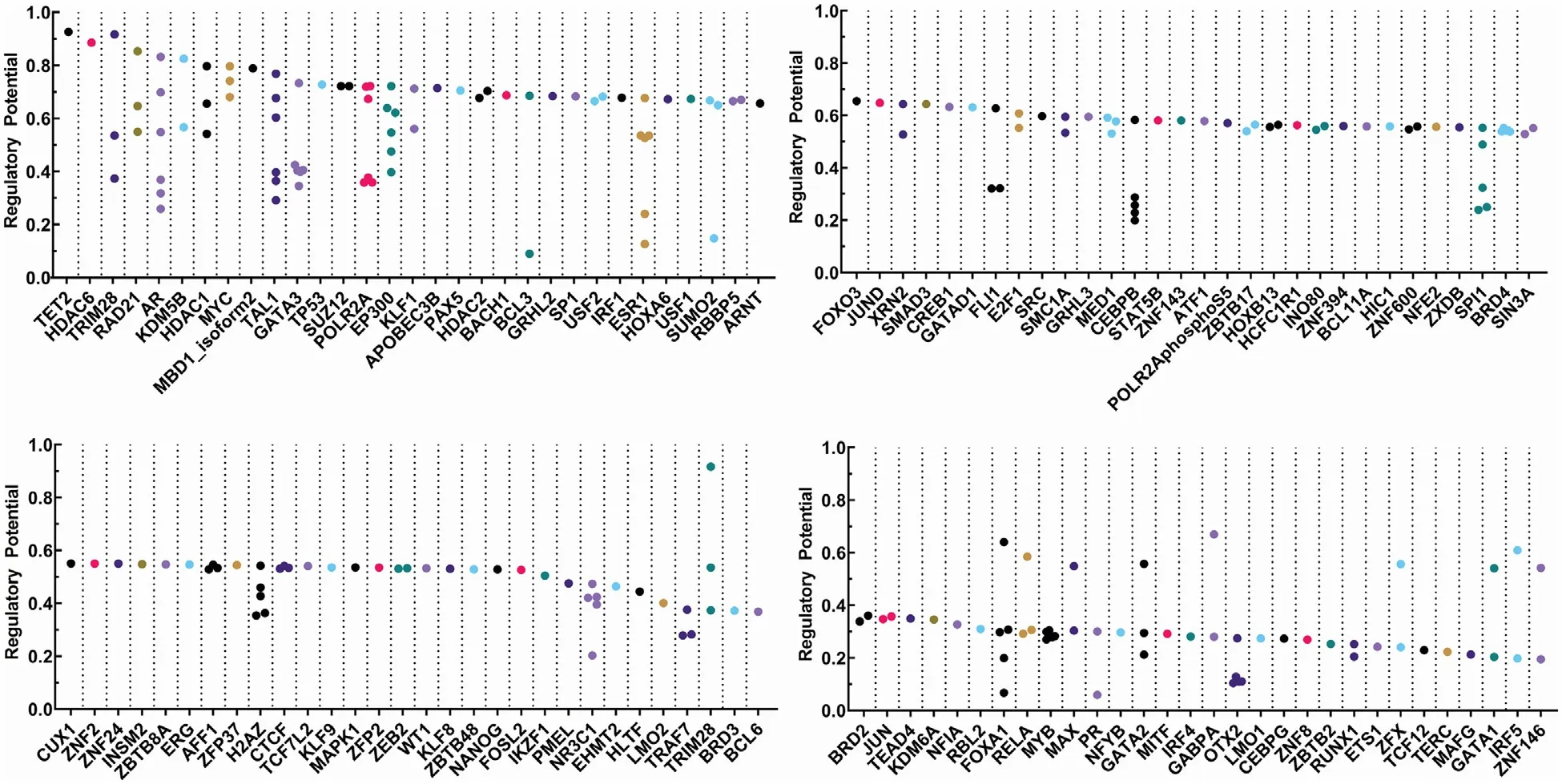
Fig.3–Transcription factors that have been identified by bioinformatic analysis which are likely to be involved in the transcription of ABCG2 .To identify possible transcription factors (TFs) involved in regulation of ABCG2, we utilized Cistrome Data Browser (DB) to acquire publicly available ChIP-Seq,DNase-Seq,and ATAC-Seq data [61,62].Cistrome DB depicted the likelihood of TFs to be involved in ABCG2 regulation via regulatory potential,on a scale of 0–1,that was derived from the Binding and Expression Target Analysis (BETA) algorithm [63];the higher the regulatory potential for a given TF,the more likely is the involvement of that TF in the regulation of ABCG2.We selected the TFs with a cutoff at > 0.2 for the regulatory potential.
As gouty arthritis affects millions of people,several drugs have been developed to treat this disease [35–37].They fall into three categories:inhibitors of xanthine oxidase (e.g.,Allopurinol),inhibitors of uric acid reabsorption in kidney(e.g.,Probenecid),and recombinant uricase to degrade uric acid into allantoin (e.g.,Pegloticase).Do we need additional drugs to treat this disease? Of course,we do! The inhibitors of xanthine oxidase and renal reabsorption cause liver injury while the recombinant uricase requires intravenous administration.Therefore,new drugs which are orally active and function via mechanisms different from those of the currently used drugs would be a welcome addition to the drug armamentarium for patients suffering from gouty arthritis.ABCG2 could be an ideal therapeutic target for development of such new drugs with a hitherto unexplored mechanistic approach to promote uric acid elimination from the body.This transporter functions in the removal of uric acid in the intestine,kidney,and liver,with intestine as the major site of action.ABCG2 contributes at least 40% to total wholebody elimination of uric acid;pharmacologic induction of this transporter could further increase the clearance of uric acid from the body and reduce the levels of this metabolite in circulation.The significant contribution of ABCG2 in uric acid elimination is clearly evident from the findings that loss-of-function mutations in this transporter cause gout in humans and thatAbcg2-null mice show defective uric acid clearance from the body resulting in hyperuricemia.ABCG2 as a therapeutic target would be effective not only in patients with normal ABCG2 but also in patients with goutassociated mutations in ABCG2.There are two variants in ABCG2 that are commonly associated with hyperuricemia and gout:Q126X and Q141K [34,38–41].The Q126X variant,which is truncated with the loss of a major portion of the protein,is non-functional whereas the Q141K variant is transportcompetent but with only~50% activity compared to wild type transporter [31].A significant number of individuals with European ancestry harbor the Q141K mutation with the carrier frequency of 11% [38].Homozygosity for this mutation is associated with hyperuricemia and gout.The Q126X variant is less prevalent compared with the Q141K variant;nonetheless,this variant is also associated with increased risk of hyperuricemia and gout.
Promotion of uric acid excretion as a therapeutic strategy for gouty arthritis could be achieved by induction of ABCG2 in intestinal tract,kidney,and liver,particularly in small intestine where uric acid production is most robust.This can be done with transcriptional activators of theABCG2gene.The promoter of this gene contains cis-acting elements for binding of numerous transcription factors (Fig.2).Most of these elements have been mapped to regions proximal as well as distal to the putative transcription start site[42].ABCG2 expression is also regulated epigenetically by promoter methylation,with hypermethylation leading to silencing of expression.Several drugs have been shown to induce ABCG2 expression in mammalian cells,and these inducers are predominantly chemotherapeutic agents.This is understandable because ABCG2 is a drug exporter and is related to multidrug resistance;when cells get exposed to certain chemotherapeutics,the cells become resistant to such drugs in due course by upregulation of ABCG2 to facilitate the removal of the drugs.But whether any of these inducers are ideal for treatment of gouty arthritis is an open question because most chemotherapeutic drugs have multiple unwanted side effects.A careful analysis of these drugs and their molecular targets,we believe that at least four transcription factors could be targeted for inducing ABCG2 expression,namely PPARγ,PPARα,NRF2,and AhR because FDA-approved drugs are available as activators of these transcription factors.If any of them could be demonstrated as potent inducers of ABCG2 in humans,such drugs could potentially be evaluated for their efficacy to treat gouty arthritis.
PPARγis a nuclear receptor that serves as the molecular target for thiazoledinediones in treatment of Type 2 diabetes[43].Pharmacologic activation of PPARγimproves insulin sensitivity in diabetic patients.Pioglitazone and Rosiglitazone are currently approved for management of hyperglycemia in Type 2 diabetes.Agonists of PPARγhave been shown to induce ABCG2 in certain cell types [44].PPARαagonists,known as fibrates,are also used for treatment of Type 2 diabetes;these drugs work by correcting dyslipidemia in diabetic patients [43].Clofibrate and gemfibrozil are examples in this class of drugs that are currently in clinical use.Dually active drugs which activate both PPARγand PPARα,are in development.PPARαselective agonists have been shown to induce ABCG2 in the intestine and liver [45].NRF2 acts through the antioxidant response element in target genes.As activation of this transcription factor induces antioxidant machinery,drugs that activate this transcription factor are being considered for management of a wide variety of diseases [46].Currently used drugs or nutritional supplements that are known to activate NRF2 include fumarate esters [47,48],sulphoraphane[49],and astemizole [50].ABCG2 in liver is a known target for NRF2 [51].AhR is a transcription factor that works through xenobiotic response element (XRE),and several FDA-approved drugs (e.g.,omeprazole) activate this factor [52].Recently we have shown that Carbidopa,used in combination with L -DOPA for treatment of Parkinson’s disease,is a potent activator of AhR [53].AhR agonists have been shown to induce ABCG2 [54,55].In addition to these transcription factors,pharmacologic strategies to induce ABCG2 expression via epigenetic mechanisms could also be considered.As promoter hypermethylation silencesABCG2,pharmaceuticals that prevent DNA methylation might prove to be useful in this regard.There are FDA-approved drugs,mostly anticancer drugs,that function as inhibitors of DNA methyltransferases(e.g.,decitabine,azacitidine).Certain nutraceuticals also have the ability to inhibit these DNA methylating enzymes.Two such compounds are Laccaic acid,a natural food additive,and epigallocatechin-3-gallate,a component in green tea,which inhibit DNA methyltransferase 1 at low micromolar concentrations [56,57].
Pharmacologic induction of ABCG2 would be effective for treatment of gouty arthritis in patients who harbor mutations in the protein that compromise the transport function.The normal transporter could also serve as a pharmacologic target for development of anti-gout drugs.If the transport function of ABCG2 can be activated with small molecules to enhance uric acid excretion,such compounds could have potential in the treatment of gouty arthritis.In this regard,it is important to note that the crystal structure of human ABCG2 has been deduced recently [58–60].The detailed structural data now available on this transporter can be exploited in the design of small molecules that could bind the transporter and activate its transport function.
10.Conclusions
ABCG2,a plasma membrane transporter involved in the export of various endogenous and exogenous metabolites and pharmaceuticals from the cells,is responsible for a significant fraction of uric acid excretion from the body.The transporter mediates the removal of uric acid in the intestine,kidney,and liver.The quantitative significance of its involvement as an important determinant of plasma levels of uric acid is evident from the findings that lossof-function mutations in this single gene are sufficient to cause hyperuricemia and gout.Therefore,induction of this transporter by pharmacological means could offer a new therapeutic strategy for the treatment of gouty arthritis.Obviously,this approach would not work in patients in whom the mutations in ABCG2 result in total loss of transport activity because induction of the inactive transporter would not have any consequence in terms of uric acid excretion.However,for patients with hyperuricemia who have either normal ABCG2 or mutant ABCG2 with only a partial loss of transport function,this approach could have therapeutic benefit.There are several FDA-approved drugs that induce this transporter(Table 2);such drugs could be re-purposed as a novel class of drugs for treatment of gouty arthritis.
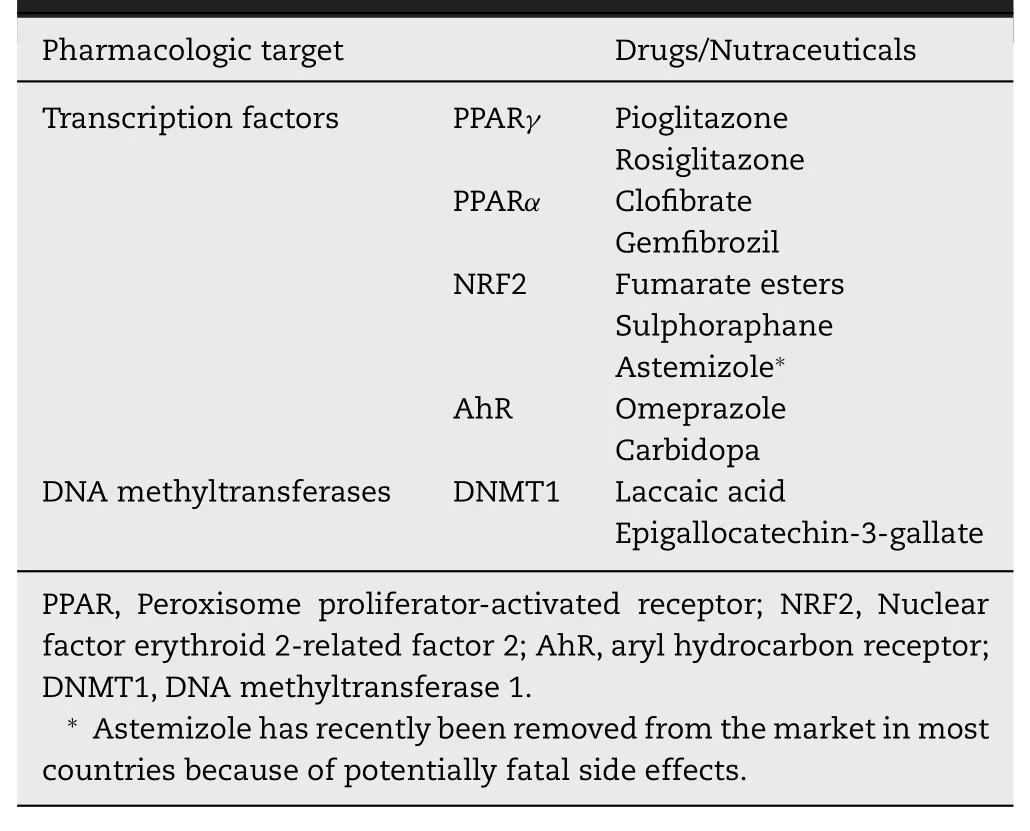
Table 2–FDA-approved drugs and nutraceuticals with potential for treatment of gout via induction of ABCG2 expression.
Conflicts of interest
The authors declare that there is no conflicts of interest.
Acknowledgments
This work was supported by the National Institutes of Health grant R41 AR074854 and the Welch Endowed Chair in Biochemistry,Grant No.BI-0028,at Texas Tech University Health Sciences Center.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.ajps.2019.10.002 .
 Asian Journal of Pharmacentical Sciences2020年2期
Asian Journal of Pharmacentical Sciences2020年2期
- Asian Journal of Pharmacentical Sciences的其它文章
- The solute carrier transporters and the brain:Physiological and pharmacological implications
- The role of transporters in cancer redox homeostasis and cross-talk with nanomedicines
- Intestinal OCTN2-and MCT1-targeted drug delivery to improve oral bioavailability
- Stimulatory effect on the transport mediated by organic anion transporting polypeptide 2B1
- Amino acid transporters:Emerging roles in drug delivery for tumor-targeting therapy
- Glutamine transporters as pharmacological targets:From function to drug design