
Intestinal OCTN2-and MCT1-targeted drug delivery to improve oral bioavailability
2020-05-15 09:35∗
∗
a Zhuang Yao Medicine Center of Engineering and Technology, Guang Xi University of Chinese Medicine, Nanning 530200, China
b School of Pharmacy, Guang Xi University of Chinese Medicine, Nanning 530200, China
c School of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, China
Keywords: Carnitine/organic cation transporter 2 (OCTN2)Monocarboxylate transporter protein 1 (MCT1)Transporter-targeting Nanoparticle Prodrug
ABSTRACT Various drug transporters are widely expressed throughout the intestine and play important roles in absorbing nutrients and drugs,thus providing high quality targets for the design of prodrugs or nanoparticles to facilitate oral drug delivery.In particular,intestinal carnitine/organic cation transporter 2 (OCTN2) and mono-carboxylate transporter protein 1(MCT1) possess high transport capacities and complementary distributions.Therefore,we outline recent developments in transporter-targeted oral drug delivery with regard to the OCTN2 and MCT1 proteins in this review.First,basic information of the two transporters is reviewed,including their topological structures,characteristics and functions,expression and key features of their substrates.Furthermore,progress in transporter-targeting prodrugs and nanoparticles to increase oral drug delivery is discussed,including improvements in the oral absorption of anti-inflammatory drugs,antiepileptic drugs and anticancer drugs.Finally,the potential of a dual transporter-targeting strategy is discussed.
1.Introduction
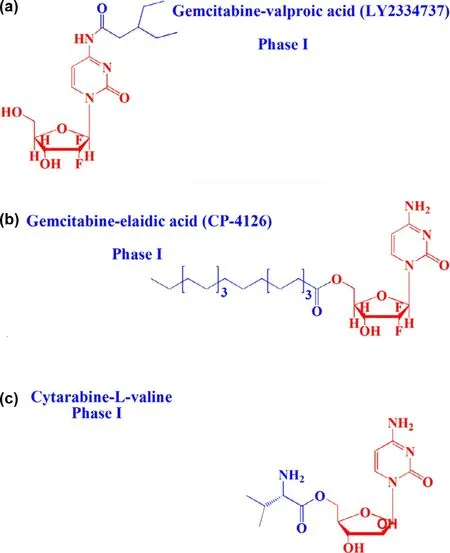
Fig.1–Chemical structures of lipophilic prodrugs and carrier prodrugs in clinical trials:a:LY2334737,b:CP-4126 and c:Cytarabine-L -valine.
Oral drug delivery has long been considered a safe and convenient route for drug development.Unfortunately,not all drugs can be administered orally.Many factors,such as stability,solubility and permeability,affect oral bioavailability [1,2].For example,oral insulin is of great significance to people with diabetes,but poor gastrointestinal tract (GI) stability leads to ineffectiveness [3].In response,lipophilic prodrugs have been designed to overcome the barriers of oral drug delivery.Zhang et al.synthesized a valproic acid ester of gemcitabine (LY2334737,phase I) to treat solid tumors [4–6].The prodrug had excellent stability in the GI,thus increasing the oral bioavailability by 1.4-fold compared to gemcitabine in rats (Fig.1 a).In addition,elaidic acid ester of gemcitabine greatly reduced tumor resistance originating from nucleoside transporterdependent cellular uptake of gemcitabine [7,8](Fig.1 b).These results revealed that lipophilic prodrug is an effective strategy to enhance oral absorption.However,the clinical results were far from satisfactory due to the high distribution on the membrane of epithelial cells and rapid degradation in the GI [9].Additionally,nanoparticle-based drug delivery systems (nano-DDS) have been used to improve the oral bioavailability of drugs,with advantages of improved drug membrane permeability,stability and therapeutic efficacy[10–12].However,the serious systemic toxicity of conventional nano-DDS caused by poor tissue selectivity significantly limits their clinical application [13,14].By contrast,receptor-targeted nano-DDS have shown significant advantages.However,the variability and heterogeneity of membrane receptors limits their targeting efficacy [15–17].Therefore,designing a highefficiency delivery system to transport drugs with poor oral bioavailability has long been a focus of pharmacists.
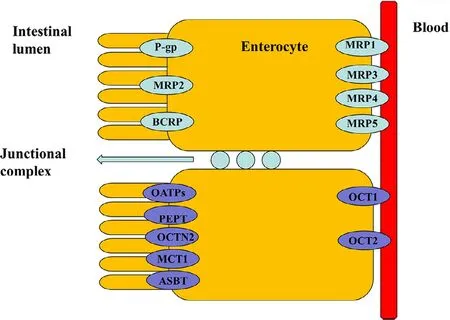
Fig.2–Diagram of major drug transporter proteins expressed by the intestinal epithelia,including intestinal uptake (purple) and efflux (green) transporters.Multidrug resistance protein (MDR1,P-glycoprotein),multidrug resistance associated protein (MRP),breast cancer resistance protein (BCRP),monocarboxylate transporter protein (MCT),peptide transporter protein (PEPT),organic anion transporting polypeptide (OATP),organic cation transporter (OCT),carnitine/organic cation transporter 2(OCTN2),and bile acid transporter (ASBT) (Reproduced with permission from [23].Copyright 2012 Elsevier B.V.).
The human body consists of several physiological barriers that express a number of membrane transporters that are categorized into two families:solute carriers (SLC) and ATP-binding cassette (ABC) [18–23].Most ABC proteins play an important role in efflux of xenobiotics out of cells to sustain cell survival,and thus providing a target for inhibitor designing for disease treatment or drug delivery[24].Meanwhile,SLC transporters transport nutrients as well as determine drug absorption and distribution.Therefore,a variety of transporter assays are development to test drug transporter interactions,transporter-mediated drug-drug interactions,and transporter-mediated toxicity [25].Recently,intestinal nutritional transporter-based drug delivery strategies have attracted more attention (Fig.2).In this strategy,the nutritional substrate is often coupled with parent drug or polymer materials to synthesize targeted prodrugs or nanoparticle-loading drugs.For instance,PEPT1-targeted prodrugs,such as zadovudine [26],acyclovir [26],ganciclovir[27],levovirin [28],and didanosine [29],showed significantly increased oral bioavailability of parent drugs.In particular,L -valine ester of cytarabine has entered into clinical evaluation[30](Fig.1 c).In addition,bile acid transporter (ASBT) is highly expressed in the small intestine and has been used as a target to develop nanoparticles for oral-transporting insulin [31].Recently,dipeptide-modified nanoparticles that target PEPT1 were proven to be a promising platform for the oral delivery of hydrophobic drugs [32].By contrast,intestinal OCTN2 and MCT1 proteins have attracted attention due to their improved transport capacity compared with PEPT1 and ASBT proteins.OCTN2 is an integral membrane protein that absorbs carnitine in the small intestine.MCT1 is also located in the intestine and is known to facilitate the transfer of mono-carboxylates from the lumen into the circulatory system [33,34].The wide distribution of the two transporters throughout the intestine provides a new possibility for designing an oral drug delivery system.Thus,substrates could be coupled to parent drugs or polymers to synthesize targeted prodrugs or nanoparticleloading drugs that are recognized by OCTN2 or MCT1 protein[33,35–38].Interestingly,we found that the transport activity of OCTN2 was higher in the duodenum and jejunum,while that of MCT1 was higher in the colon and rectum,although both proteins were expressed throughout the intestine.If a prodrug or nanoparticle was designed to target two transporters simultaneously,it would improve oral bioavailability compared with a mono-transporter targeted strategy.Accordingly,the present article summarizes development of the intestinal OCTN2 and MCT1 protein-targeted strategy.First,some basic information of the transporter,such as topological structure,characteristics and function,expression and key features of substrates,are reviewed in this article.Second,studies of prodrug-and nanoparticle-targeted intestinal OCNT2 and MCT1 protein are reviewed.Finally,a dual transporter-based drug targeted strategy is also discussed.
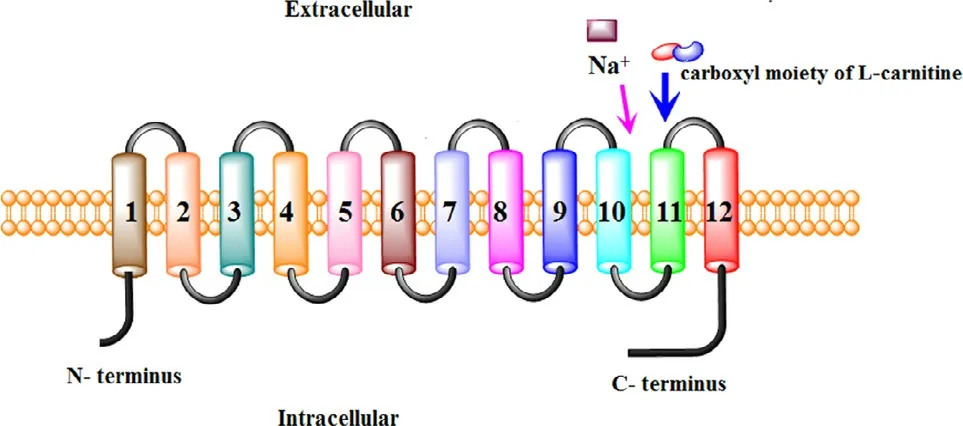
Fig.3–Proposed membrane topology of OCTN2(Reproduced with permission from [31].copyright 2008 American Chemical Society).
2.OCTN2 transporter
2.1.Topological structure of OCTN2
Obtaining the topological structure of OCTN2,especially the orientation of the amino (N-) and carboxyl (C-) termini of a trans-membrane section,the number of TMD,and the position of the trans-membrane segments in the protein sequence,may contribute to revealing protein-substrate interactions and clarify the important biological functions of proteins [39].To this end,hydropathy profiles were employed,and the results showed that OCTN2 possessed 12 TMD,with the intracellular C-terminus and N-terminus exposed to the cytosol (Fig.3) [40].To affirm the precise domain of the substrate binding site,site-directed mutagenesis was performed.Wang et al.found that the mutation of amino acid E452K,located in the hydrophilic loop between TMD 10 and 11,resulted in decreased affinity for Na+but not carnitine [41].Thus,this segment was a Na+binding site.Moreover,Seth et al.reported that the mutation of S467C and P478L,located in TMD 11 in OCTN2,decreased the affinity of anions [42].Therefore,this segment was a carboxyl moiety binding site of carnitine.Seth et al.also found that carnitine uptake was inhibited by TEA in the mutation model,whereas TEA did not show complete competitive inhibition for carnitine uptake,suggesting that the binding sites for TEA and carnitine partially overlap on OCTN2 [42].However,the binding site of cations in OCTN2 has not been reported until now.
2.2.Characteristics and function of OCTN2
L-carnitine is an essential nutrient with various important physiological functions:(i) it is vital forβ-oxidation of longchain fatty acids in the mitochondria;(ii) it maintains cell membrane stability by acetylating membrane phospholipids;and (iii) it protects cells in metabolic disorders by antioxidant effects [43–46].In mammals,L -carnitine is absorbed from the small intestine by OCTN2 [47].With its help,the concentration of intracellular carnitine is approximately 50 times higher than that in the extracellular space,indicating that OCTN2 is a protein with high transport capacity [48].Only a few organs,such as the liver and kidneys,have the ability to biosynthesize L -carnitine [49].Therefore,many tissues,such as skeletal and heart muscle,are highly dependent on OCTN2 protein for supplying L -carnitine.
OCTN2 is a specific Na+/carnitine co-transporter and contributes to carnitine homeostasis in humans.OCNT2 has been cloned from rat [50,51],human [52]and mouse [53].The Michaelis constant of carnitine transport by OCTN2 is in the range of 10–20 μM,indicating that OCTN2 is a high affinity transporter.OCTN2 also transports drugs,such as TEA,ipratropium,prednisolone,and beta-lactam antibiotics[54–56],in a pH-dependent manner.Therefore,the cellular uptake of carnitine would inevitably be inhibited when taking ionic medicine,such as cephaloridine,levofloxacin and grepafloxacin,resulting in carnitine deficiency [57,58].Although several reports have focused on drug-drug interactions mediated by OCTN2,few studies have provided key data,such as IC50/KI,which are important for drug development.Thus,we can utilize OCTN2 protein as a target for oral prodrugs or nanoparticle design.
2.3.The expression of OCTN2
OCTN2 transporters are widely distributed in various organs in human and mouse (Table 1) [59,60].In humans,it is found in the liver,heart,placenta and muscle [61].Recently,OCTN2 was discovered on hematopoietic cells [62].The expression of OCTN2 can be regulated by some pathological conditions.For instance,intestinal OCTN2 was downregulated in inflammatory bowel disease (IBD) [63].The mRNA level of OCTN2 was significantly reduced in dilated nonischemic cardiomyopathy (DCM) patients,while it was un-affected in patients with acute myocarditis (AMC) [64].Additionally,therapeutic drugs also affect the expression of OCTN2 in tumors.Due to the high expression of proliferator-activated receptor alpha (PPARA) and gamma (PPARG),the expression of OCTN2 in tumors is downregulated.However,the decreased level of OCTN2 in tumor cells could be restored by decitabine,a demethylating reagent,resulting in the increased uptake of oxaliplatin [65].
In this article,we focus on the expression of OCNT2 in the intestine.Kato found that OCTN2 is mainly localized at the brush-border of apical membranes of intestinal epithelial cells [66].By detecting the mRNA level of different intestinal segments,Meiery found that OCTN2 was expressed throughout the intestine,while PepT1 is only expressed in the anterior of the small intestine.Theseresults indicate that the interaction time and absorptive area of substrate to transporter is greatly increased,which is suitable for transporter-based prodrug and nanoparticle design [67].

Table 1–Characteristics of the OCTN2 transporter (Reproduced with permission from [59].Copyright 2012 John Wiley &Sons,Ltd.).
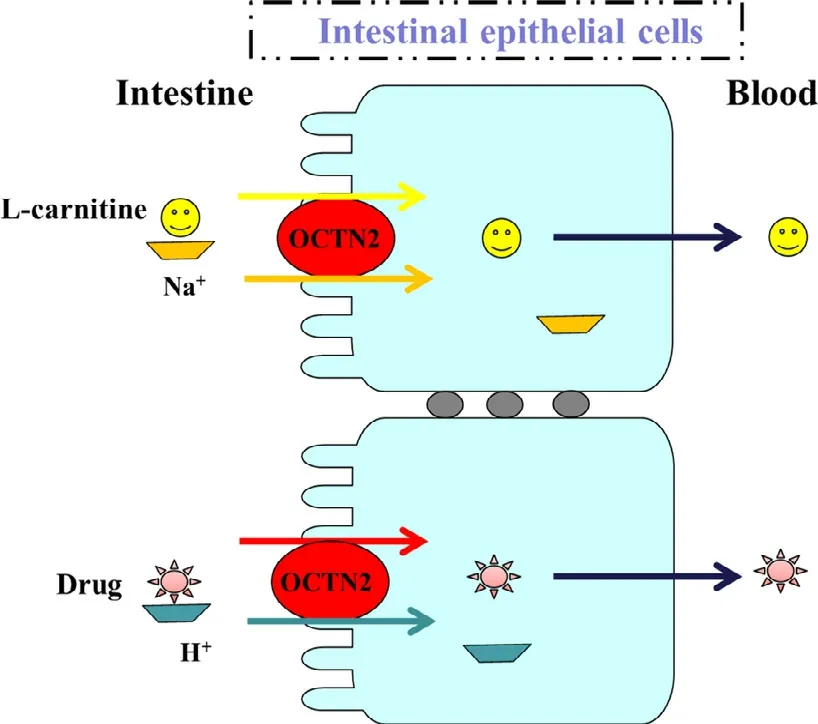
Fig.4–Model of OCTN2-mediated transport of L -carnitine and drugs in intestinal epithelial cells.Carnitine is taken up by cells in a sodium ion-dependent manner,while drug transport is sodium ion-independent.Carnitine binding site is only functional when sodium ion is present on the same side.Carnitine/organic cation transporter 2 (OCTN2)(Reproduced with permission from [66].Copyright 2006 American Society for Pharmacology and Experimental Therapeutics).
2.4.Key features of OCTN2 substrates
OCTN2 preferentially transports L -carnitine and cations,but the transportation mechanism is different.As shown in Fig.2,the transport of L -carnitine is Na+-dependent,while the transport of cations is H+-dependent [68,69].Interestingly,the L -carnitine transported out of cells is independent of OCTN2.In addition to TEA and L -carnitine,OCTN2 also accepts valproic acid.Therefore,Ohashi et al.proposed a model of substrate binding sites of OCTN2 protein,including a cation site,anion site and Na+site (Fig.4) [69].
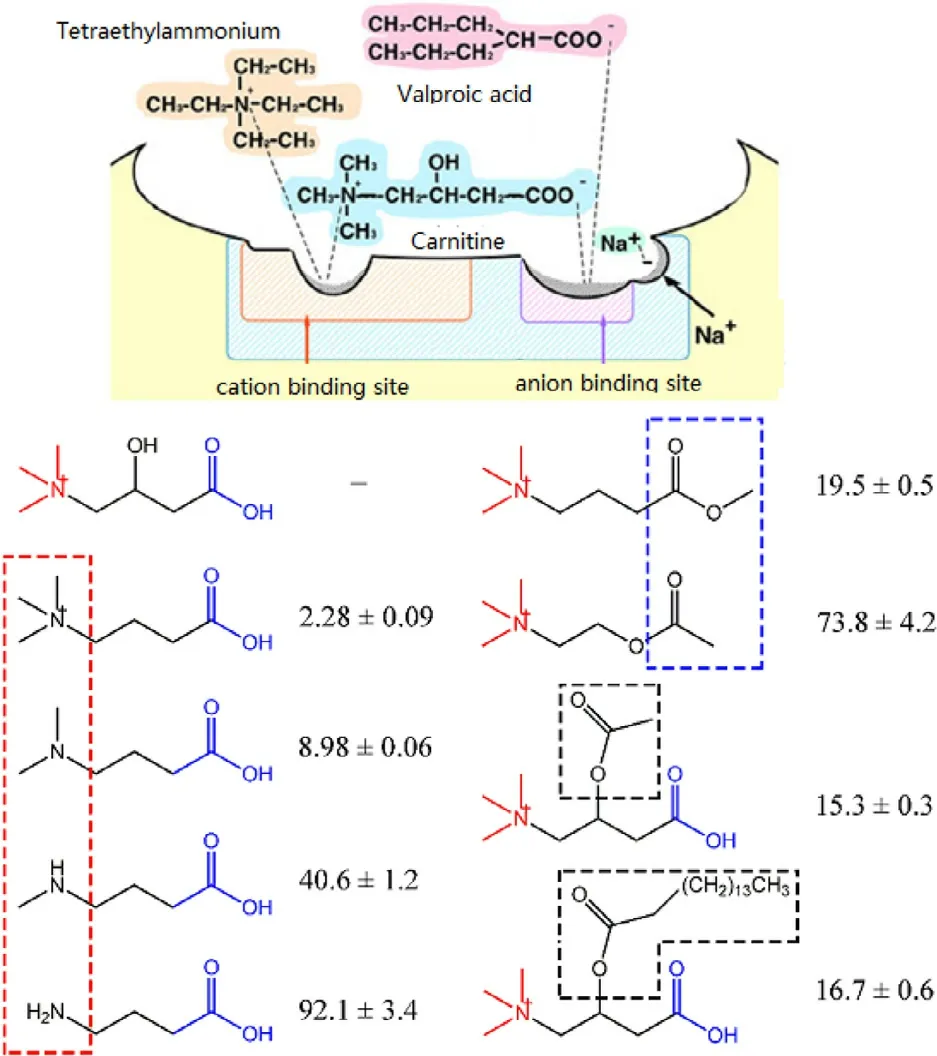
Fig.5–Effects of L -carnitine analogues on OCTN2-mediated L -carnitine transport.The values given are from competition experiments and show the percent of L -carnitine (2.5 μM) uptake when measured in the presence of the analogues;the concentration of L -carnitine analogues is 500 μM,and that of the two 3 ′–hydroxyl derivatives is 100 μM.L -carnitine uptake measured in the absence of any competitors is taken as 100%.carnitine/organic cation transporter 2 (OCTN2) (Reproduced with permission from [70].Copyright 2011 Wiley-Liss,Inc.).
To optimize the derivative position of L -carnitine for prodrug design,22 analogues were used [70].The results suggested that the affinity of substrate for OCTN2 protein decreased when L -carnitine was esterified at the carboxylic position.Additionally,the affinity of substrate for OCTN2 also depends on the type of amine of L -carnitine.The binding ability follows the order of quaternary amine>tertiary amine>secondary amine>primary amine (Fig.5).Accordingly,the amine group and carboxylate group of the L -carnitine are critical groups in the recognition of OCTN2 for substrates.By contrast,the 3′-OH shows a negligible effect and thus should be used as conjugate site for prodrug design.In response,L -carnitine was covalently coupled to prednisolone through the 3′-OH (PDSC) and carboxylate group(PDC) to synthesize OCTN2-targeted prodrugs.PDSC displayed a 1.79-fold increase in OCNT2-mediated cellular uptake than prednisolone,proving that 3′-OH can be used as a derivative site [56].
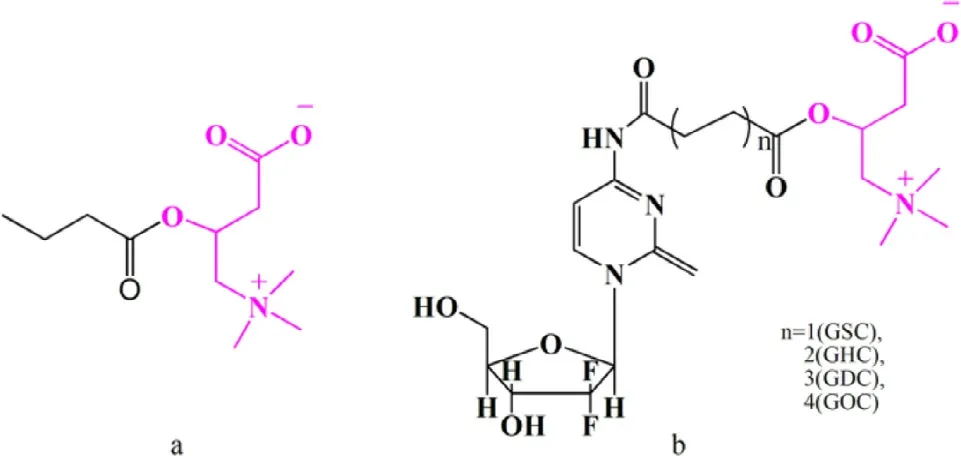
Fig.6–L -carnitine conjugated prodrugs for OCTN2 targeting.L -carnitine is identified in pink,and the parent drug is identified in black.a:Butyrate ester of L -carnitine;b:L -carnitine ester of gemcitabine.GSC:L -carnitine-succinic-gemcitabine,GHC:L -carnitine-hexylic-gemcitabine,GOC:L -carnitine-octanedioic-gemcitabine,GDC:L -carnitine-decanedioic-gemcitabine.carnitine/organic cation transporter 2 (OCTN2).
2.5.Strategies to target OCTN2 for enhanced oral drug delivery
Despite many attempts,the current oral strategies are still far from satisfactory.Due to progress in molecular biology,many SLC transporters have been discovered in the intestine.The typical transporters are PEPT1,ASBT and OCTN2,which can accept both nutrients and therapeutic drugs.The molecular revolution of membrane transporters has spurred novel strategies to design transporter-targeted prodrugs and nanoparticles for increased intestinal permeability of drugs.Recently,great progress has been made,which will be discussed in the subsequent sections.
2.5.1.OCTN2-targetedoralprodrugs
Transporter-targeted prodrug strategies have obvious advantages,such as high permeability and appropriate water solubility,in which pro-moieties as ligands are covalently attached to drugs to selectively target certain membrane transporters.For example,OCTN2 protein was used as a target to design oral prodrugs for butyrate [63]and gemcitabine [33](Fig.6).Butyrate is a short-chain fatty,with a role in preventing ulcerative colitis.Butyrate is mainly absorbed via intestinal MCT1 protein [71–75].However,its expression is downregulated in the intestinal mucosa of patients with IBD [76].Additionally,L -carnitine has been shown to have protective effects in ulcerative colitis [77,78].Accordingly,a butyrate ester of L -carnitine was synthesized by Srinivas et al.to treat gut inflammation (Fig.6 a).Butyrate ester of L -carnitine is taken up through OCTN2 or ATB0+and then hydrolyzed to release butyrate and L -carnitine in epithelial cells for the treatment of gut inflammation.The cellular uptake experiment was performed in ATB0+high expressing oocytes and hOCTN2-transfected HRPE cells.The results demonstrated that the butyrate ester of L -carnitine inhibited L -carnitine (IC 50=0.3 μM) and glycine (IC 50=4.6 mM) uptake,indicating that prodrug was a substrate of ATB0+and OCTN2.Moreover,the uptake results of butyrate ester of L -carnitine in two cells showed different levels,suggesting that OCTN2 is a high affinity transporter (K0.5=0.04 μM),whereas ATB0+is a low affinity transporter (K0.5=1.4 mM).
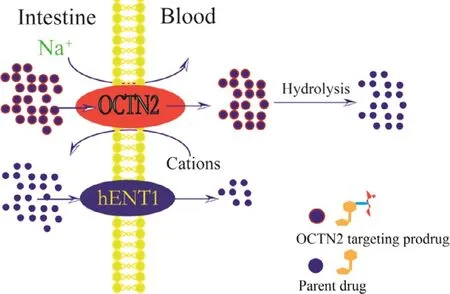
Fig.7–Mechanism of OCTN2-targeting prodrug uptake by the intestine.carnitine/organic cation transporter 2(OCTN2),hENT1:human equilibrative nucleoside transporter-1.
Gemcitabine has been used as a first line drug in many solid tumors for chemotherapy,including pancreatic and nonsmall cell lung cancer [79–81].However,hematological toxicity and other side effects significantly limit its clinical application by the intravenous route [6].To develop high-efficacy,low-GI toxicity oral gemcitabine products,we proposed an OCTN2-targeting oral prodrug strategy.Four targeted prodrugs,differing in the length of linkage,were synthesized,as shown in Fig.6 b.L -carnitine was used as the ligand and was coupled with the N4-amino of gemcitabine.Compared to gemcitabine,the prodrugs displayed significantly improved stability(3-fold) and oral bioavailability (5-fold).Further,the cellular uptake of the prodrugs was Na+-dependent and temperaturedependent and was inhibited by L -carnitine.These results indicate that the prodrugs were absorbed into cells via OCTN2.In addition,a molecular docking study was performed to evaluate the affinity of the prodrugs for OCTN2.The hexane diacid-linked prodrug exhibited the highest affinity for OCTN2 among the prodrugs.Importantly,safety studies using hematoxylin and eosin (H&E) histological analysis suggested that there was no toxicity of the targeted prodrugs in mice at short exposure durations to the heart,liver,spleen,lung or kidney,likely due to low tissue distribution and rapid clearance in various tissues.These results suggest that L -carnitine ester of gemcitabine is a promising strategy to enhance the oral bioavailability of gemcitabine(Fig.7).
PEPT1 is a low affinity transporter with largeKmandVmax,indicating that this transporter is not easily saturated under high concentrations of substrate and thus is an ideal target for development of an oral prodrug [82].By contrast,OCTN2 is a transporter with lowKmandVmax.Thus,it is easily saturated and is theoretically not suitable for the design of prodrugs.Amazingly,OCTN2 showed better transport capacity compared to PEPT1 in the above studies,likely due to its fast transport speed.Therefore,OCTN2 is also a good target for the design of oral prodrugs.
2.5.2.OCTN2-targetedoralnanoparticles
Although the transporter-targeted prodrug strategy has made great progress,poor stability and low activatable efficiency still limit its clinical application [18,20].By contrast,transporter-based nano-DDS has shown obvious advantages in oral drug delivery,such as improved drug stability,permeability and safety.Paclitaxel (PTX) is widely used in the clinic for treating solid tumors [83].However,no oral preparations have been developed for marketing due to its poor solubility and high GI toxicity.Based on the promise of OCTN2 as a target for oral drug delivery,we attempted to design and synthesize OCTN2-targeting PLGA nanoparticles loading paclitaxel [34].All nanoparticles showed slower but prolonged release of paclitaxel compared to free taxol.L -carnitine-modified nanoparticles showed increased cellular uptake compared to non-targeted nanoparticles and was inhibited by L -carnitine,revealing that OCTN2 helps to increase uptake of the nanoparticles.Further,we evaluated the effect of Na+and Cl−on the binding of OCTN2-targeted nanoparticles.The results showed that the binding of OCTN2-targeted nanoparticles was only dependent on Na+.However,these results could simply reveal the binding of nanoparticles to OCTN2 protein and not reflect the actual uptake mechanism since the size of the nanoparticle is larger than free L -carnitine by several orders of magnitude.Therefore,we further evaluated the endocytosis mechanism of the nanoparticle using various inhibitors,including indomethacin(an inhibitor of caveolin),chlorpromazine (an inhibitor of clathrin),colchicine (an inhibitor of macropinocytosis) and quercetin (an inhibitor of caveolin/clathrin).The results indicated that nanoparticles enter the cell via clathrin-and caveolae-mediated endocytosis.Accordingly,the specific transport process of OCTN2-targeted nanoparticles is summarized as follows (Fig.8):(i) nanoparticles bind to OCTN2 in a Na+-dependent manner;(ii) after binding,OCTN2 transporter shifts from outward-facing to an occluded state,which induces membrane invagination;(iii) then,endocytosis occurs,with multipoint binding increasing the interaction and accelerating nanoparticle absorption;(iv) after endocytosis,some complexes may fuse with the lysosome,resulting in protein degradation,and some may go through the cytosol for transcytosis and release of nanoparticles to the basolateral side.The pharmacokinetics study exhibited that the oral bioavailability of paclitaxel loading in targeted nanoparticles increased by 3 times compared with nontargeted nanoparticles,confirming the contribution of OCTN2-targeted nanoparticles.Interestingly,thetmax of nanoparticles was three times longer than the parent drug,likely because the endocytosis process of nanoparticles is complex and time-consuming.These results suggest that OCTN2 is an ideal target for increasing the oral absorption of nanoparticle-loading drugs.
3.MCT1 transporter
3.1.Topological structure of MCT1
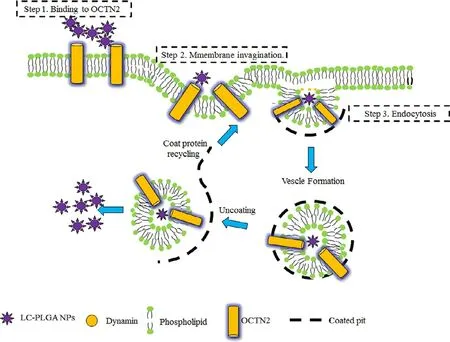
Fig.8–Mechanism of L -carnitine-conjugated nanoparticle uptake.LC-PLGA NPs bind to OCTN2 on the cell membrane in the presence of Na +,and OCTN2 then shifts conformation from outward-facing to an occulated state,forming a complex containing LC PLGA NPs,Na + and OCTN2.The formed complex induces membrane invagination and subsequent endocytosis.Additionally,a multivalent binding connection between nanoparticles and the membrane further accelerates the absorption of nanoparticles.After endocytosis,some complexes might fuse with the lysosome,resulting in protein degradation,and some may go through the cytosol for transcytosis and release of nanoparticles to the basolateral side (Reproduced with permission from [34].Copyright 2017 WILEY-VCH Verlag GmbH &Co.KGaA,Weinheim).
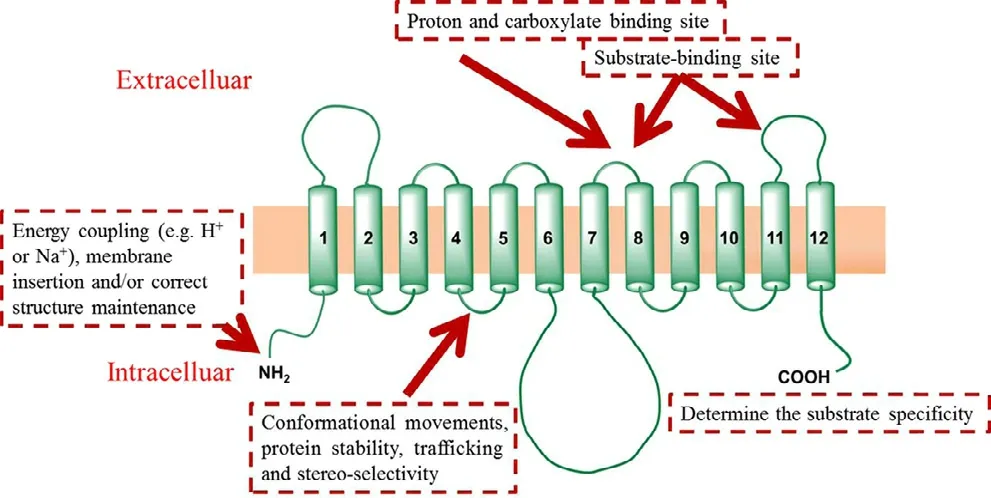
Fig.9–Proposed membrane topology of MCT1 (Reproduced with permission from [64].Copyright 2011 American Society for Investigative Pathology.).MCT1:mono-carboxylate transporter protein 1.
As shown in Fig.9,the MCT1 transporter contains 12 TMD,which are predicted by hydropathy plots [84]using the Kyte–Doolittle algorithm [85]and the TMpred program [86].To affirm the precise sites relevant to substrate binding,various technologies were used.Inhibitor and photo affinitylabeling studies suggest that the N-terminus of the protein is responsible for the energy coupling (e.g.,H+or Na+),membrane insertion and correct structure maintenance [87].Mutagenesis studies revealed that some critical domains were related to the function of MCT1.For example,TMD 8 plays an essential role in proton and carboxylate binding[88].The domain of the C-terminus of MCT1 is thought to help determine substrate specificity [89,90].The loop between TMD 7 and 8 and the loop between TMD 11 and 12 are the substrate-binding sites [91].The domain between TMD 4 and TMD 5 contribute to the conformational movements,protein stability,trafficking and stereo-selectivity [92].However,no crystal structure of MCT1 has been reported to date.
Another important factor for MCT1 is ancillary proteins with roles of proper intracellular trafficking,cell surface expression,and function.Previous studies have suggested that MCT1 protein requires the presence of CD147 or the related protein GP70,which contain one trans-membrane,two immunoglobulin and one carboxyl terminus cytoplasmic tail[93].The interaction of the carboxyl terminus cytoplasmic tail and the fluorescent protein pulls the MCT1 protein from the synthetic site to the cell surface.Therefore,loss of the co-expression of ancillary proteins would result in the accumulation of MCT1 in the endoplasmic reticulum or Golgi body [94].
3.2.Characteristics and function of MCTs
Glycolysis is very important to muscle,red blood cells and tumor cells for maintaining normal function and proliferation [95].Large amounts of lactic acid produced by glycolysis are rapidly pumped out of cells via proton-linked mono-carboxylate transporters (MCTs) [96–98].Moreover,butyrate,the principal energy source for colonic epithelial cells originating from microbial fermentation of dietary carbohydrates,is also a substrate of MCT1 protein in the intestine [99].Butyrate is present in the colon in millimolar concentrations and plays an important role in their proliferation,differentiation and apoptosis [99].Furthermore,MCTs are essential for the transport of important monocarboxylates,including pyruvate,ketone bodies acetoacetate,andβ-hydroxybutyrate.Therefore,MCTs are vital transporters for cellular metabolism and intercellular signal transduction[84,100](Fig.10).As such,MCTs have attracted increased attention.MCTs belong to the SLC16A family and contain 14 family members,MCT1-MCT14.As shown in Table 2,MCTs are widely expressed at various tissues,including the intestine,brain,kidney,liver and tumor,delivering various substrates[101].Among the MCT transporters,we focus on MCT1 in this review because it is a mono-carboxylate transporter mainly responsible for the proton-linked transport of several monocarboxylate analogues and is also a potential target for oral drug delivery.
3.3.The expression of MCT1
As shown in Fig.9,the expression of MCT1 is rather ubiquitous.In the heart,cardiomyocytes display high MCT1 expression [102–104].In the liver,it is present on hepatocytes and is responsible for pumping lactic acid out of cells[102,103,105,106].In the GI,different levels of MCT1 protein are widely distributed on the epithelial cells of the small and large intestines,contributing to the absorption of monocarboxylates from the lumen to the blood [107–110](Fig.9).Recently,a growing number of studies have shown that MCT1 protein is highly expressed in tumors to take up large amounts of lactic acid into cells as fuel for aerobic glycolysis[111].The expression of MCT1 protein can be upregulated in human muscle by exercise training to extrude lactate and H+to the interstitium.Further,the level of increased MCT1 expression is related to the intensity of training [112].In addition,cerebral ischemia can upregulate the expression of MCT1 in astrocytes for transferring intracellular lactate to the extracellular space,which facilitates lactate utilization by injured neurons to improve neurological deficits [113].In inflammatory bowel disease patients,the expression of MCT1 is strongly downregulated,leading to butyrate deficiency [76].Beyond this,butyrate,a transporter substrate,has been shown to increase MCT1 expression at the mRNA and protein levels[114].
Due to the discussed potential of MCT1 protein in developing intestinal-targeted prodrugs or nanoparticles,we highlight the expression of MCT1 in the intestine.Shimoyama et al.detected the expression of intestinal MCT1 using western blotting and immunohistochemical staining.The results showed that MCT1 protein was observed on the basolateral membranes of epithelial cells throughout the intestines.The immuno-reactivity was greater in the large intestine than in the small intestine [115].Furthermore,the mRNA of MCT1 increased by 20-fold in the large intestine compared with the small intestine [116].These data indicate that the major transport site of MCT1 protein is the posterior segment of the small intestine and large intestine.
3.4.Key features of MCT1 substrates
MCT1 is responsible for the bidirectional transport of monocarboxylates,such as pyruvic acid,lactic acid,acetoacetic acid andβ-hydroxybutyric acid,in an H+-dependent manner[117].As shown in Fig.11,the affinity for pyruvic acid is the best among the four MCT1 substrates.Interestingly,monocarboxylate transport by MCT1 is improved when the C-2 and C-3 positions are substituted [118],likely due to the increase in its binding affinity for the transporter.In addition,the transport level of L -lactic acid is higher than D -lactic acid,indicating that MCT1 has stereo-selectivity for substrate transport [117].Beyond these,some therapeutic compounds could also be transported by MCT1,includingβ-lactam antibiotics,penicillins [119],nonsteroidal anti-inflammatory drugs,valproic acid [120],atorvastatin [121]and nateglinide[122].Through structural analysis,we found that these drugs all contained a free carboxyl group.Thus,we conclude that the carboxyl group may be a vital factor for transport by MCT1.
3.5.MCT1-targeting oral prodrug
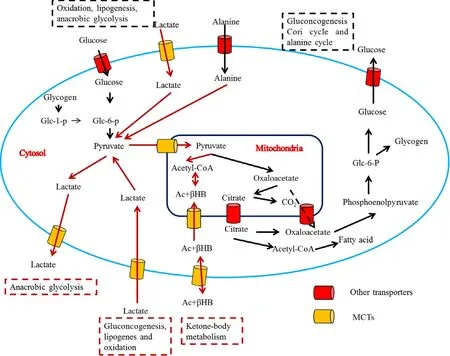
Fig.10–Metabolic pathways involving monocarboxylate transport across the mitochondrial and plasma membranes(Reproduced with permission from [84].Copyright 1999 Biochemical Society.).Glc-1-P and Glc-6-P,glucose 1-phosphate and glucose 6-phosphate;Ac -βHB,acetoacetate plus β-hydroxybutyrate.
The MCT1 transporter is overexpressed throughout the intestine and is responsible for the transport of short-chain fatty acids from the intestine to the blood.As such,MCT1 can provide a broadly oral absorption window compared with other intestinal transporters.Therefore,the MCT1 transporter has attracted increased attention for the rational design of oral prodrugs,such as gabapentin [123],5-fluorouracil[124]and gemcitabine.Gabapentin is a GABA analogue that has been used for the treatment of anxiety disorders,restless legs syndrome and hot flashes [125–127].However,low oral bioavailability,high inter-patient variability,nonlinear pharmacokinetics and short half-life have severely limited its clinical application.To overcome these shortcomings,XP13512,a novel prodrug of gabapentin,was synthesized to target intestinal MCT1 (Fig.12 a).The prodrug stability,cellular uptake and pharmacokinetics were evaluated.The stability results showed that the hydrolysis rate of XP13512 to gabapentin was slow in the plasma but rapid in liver homogenates,indicating that the primary bio-activation site is the liver followed by the plasma.To confirm the contribution of MCT1 transporter,MCT1-transfected HEK cells were used.As expected,the prodrug significantly inhibited the uptake of lactate (IC50620 μM),but gabapentin did not.Moreover,lactate decreased the cellular uptake of prodrug by 50%.These results demonstrated that the prodrug is taken up by the MCT1 transporter.The oral bioavailability of gabapentin prodrug increased from 25% to 84% in monkeys.Presently,XP13512 is currently in clinical trials for the treatment of restless legs syndrome and neuropathic pain.This research indicates that intestinal MCT1 is an ideal target for enhancing the oral absorption of gabapentin.
Although MCT1 can accept various substrates,shortchain fatty acids are the appropriate high-affinity substrates.Accordingly,those are usually selected as ligands for MCT1-targeted prodrug design.We synthesized two MCT1-targeting prodrugs using hexanedioic acid and octanedioic acid as ligands to increase the oral bioavailability of 5-fluorouracil(Fig.12 b).The prodrugs were stable in phosphate buffers(pH1.2,6.8 and 7.4) but rapidly released 5-fluorouracil in the plasma and liver homogenate,indicating that the prodrugs could be stable in the GI before interaction with MCT1 protein and were easily activated in the circulatory system.Theinsitusingle-pass intestinal perfusion experiment showed that the permeability of the prodrugs increased by 3-fold compared with 5-fluorouracil.Further,the MCT1 competitive inhibitor(quercetin) and substrate (butyrate) significantly decreased the permeability of the prodrugs.These results indicate that the MCT1 transporter is involved in the absorption of the prodrugs.To further confirm the contribution of MCT1,an uptake mechanism study was performed using Caco-2 cells.The results demonstrated that the uptake process was saturable,temperature-dependent,and MCT1-mediated.Anin vivopharmacokinetics study exhibited that the bioavailability of the prodrugs increased by 1–4-fold over 5-fluorouracil.These results further indicate that the MCT1 transporter can be used as a target to design oral prodrugs.
An ideal prodrug should be rapidly converted to the parent drug in the blood.However,little attention has been paid to this aspect during transporter-targeting prodrug design.Therefore,we put forward a dual-function MCT1-targetedprodrug strategy to overcome this problem.Di-acid monoamidation linkages with different carbon numbers were used as ligands and the immolative spacer,which were covalently linked to the N4-amino group of gemcitabine (Fig.12 c).The stability,cellular uptake mechanism,and pharmacokinetics were performed.Compared to gemcitabine,the prodrugs exhibited better MCT1 affinity,higher gastrointestinal tract stability (3-fold),improved oral bioavailability (8.8-fold),and low gastrointestinal toxicity.The uptake of the prodrugs in Caco-2 cells was significantly inhibited by 1 mM butyrate(decreased by 1.3-to 3.0-fold),while the cellular uptake of gemcitabine was not affected.In addition,the cellular uptake mechanism results exhibited that the uptake process was saturable,temperature-dependent,and MCT1-mediated.Aninvivopharmacokinetics experiment demonstrated that the prodrugs had significantly improved half-lives by 2-fold and oral bioavailability by 8.8-fold.Thus,the high membrane permeability and good gastrointestinal tract stability of the prodrugs significantly increased their pharmacokinetic behavior.It is worth noting that prodrug 2 with a 6-carbon linker exhibited the maximum oral bioavailability over the other prodrugs,indicating that the length of the diacid linkage is vital in the performance of MCT1-targeted prodrugs.Moreover,the ratio of the prodrugs to gemcitabineinvivoincreased with the extension of the carbon chain linkage,suggesting that the length of the linkage can modify the parent drug release rate.Therefore,we investigated the activation mechanism of prodrug 2 by incubation in phosphate buffers with the help of UPLC-MS/MS-Q-TOF.The molecular weight changed from 392.20612 [M Prodrug 2+H]+to 263.93022 [M Gem+H]+in pH 7.4,but the amide hydrolysis intermediate molecular weight of 409.99214 [M Prodrug 2+H2O+H]+was not observed.This result indicated that the prodrugs were activated through a cyclizationactivating pathway.The above research showed that the diacid mono-amidation linkage improves the oral absorption via MCT1 and also modifies the drug release by cyclizationactivation (Fig.13).
3.6.The MCT1-targeting oral nanoparticle
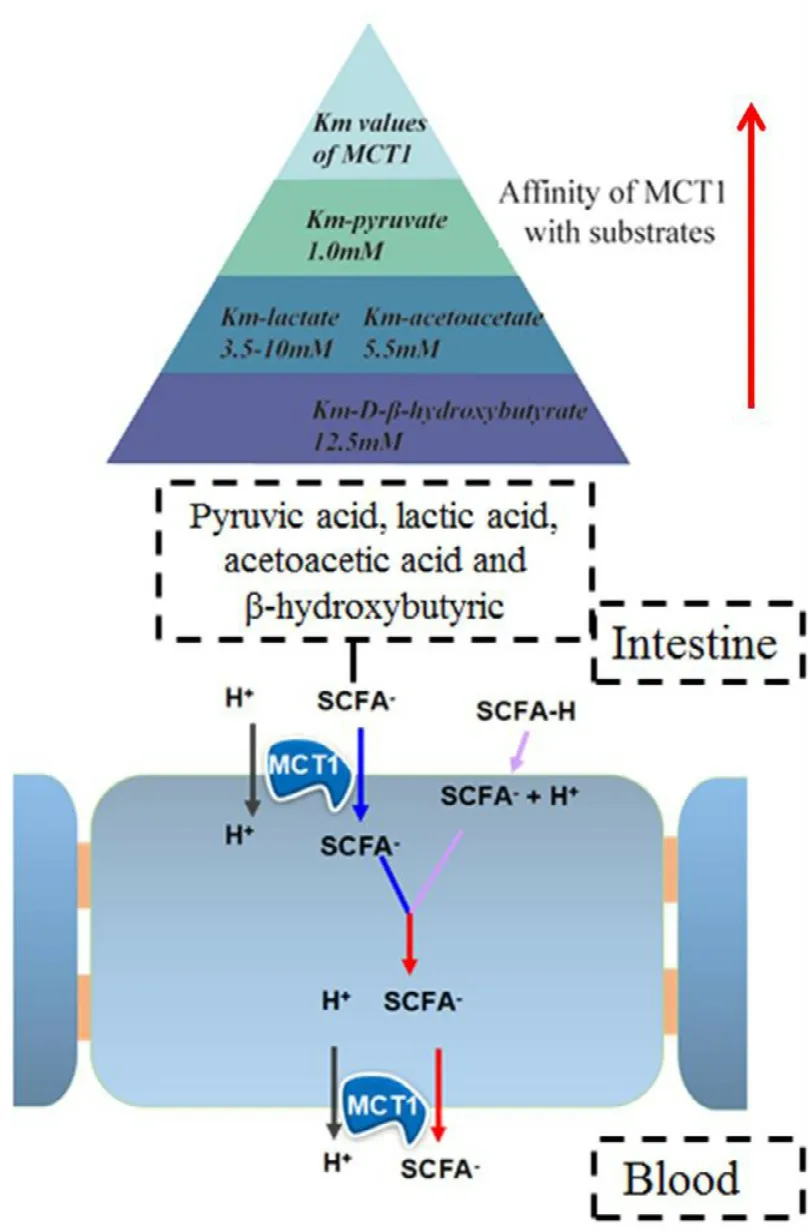
Fig.11–Uptake mechanism of substrate via MCT1 and the affinity for different substrates (pyruvate,L -lactate,acetoacetate,D -β-hydroxybutyrate).The affinity of MCT1 for the substrates was judged by the Km :the smaller the Km,the higher the affinity.MCT1:mono-carboxylate transporter protein 1.
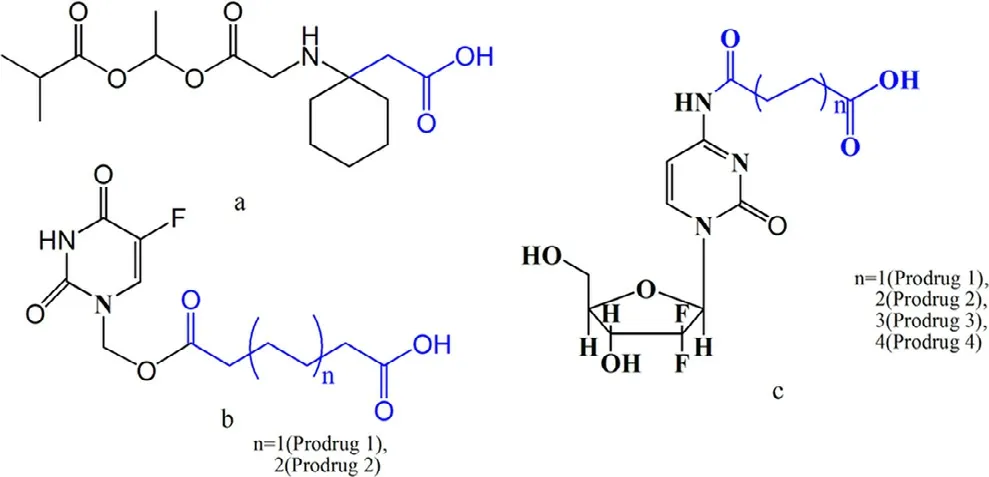
Fig.12–Fatty acid-conjugated prodrugs for MCT1 targeting.Fatty acid is identified in blue,and the parent drug is identified in black;a:XP13512;b:hexanedioic acid (n=1)and octanedioic acid (n=2) ester of 5-fluorouracil;c:di-acid mono-amidation of gemcitabine.
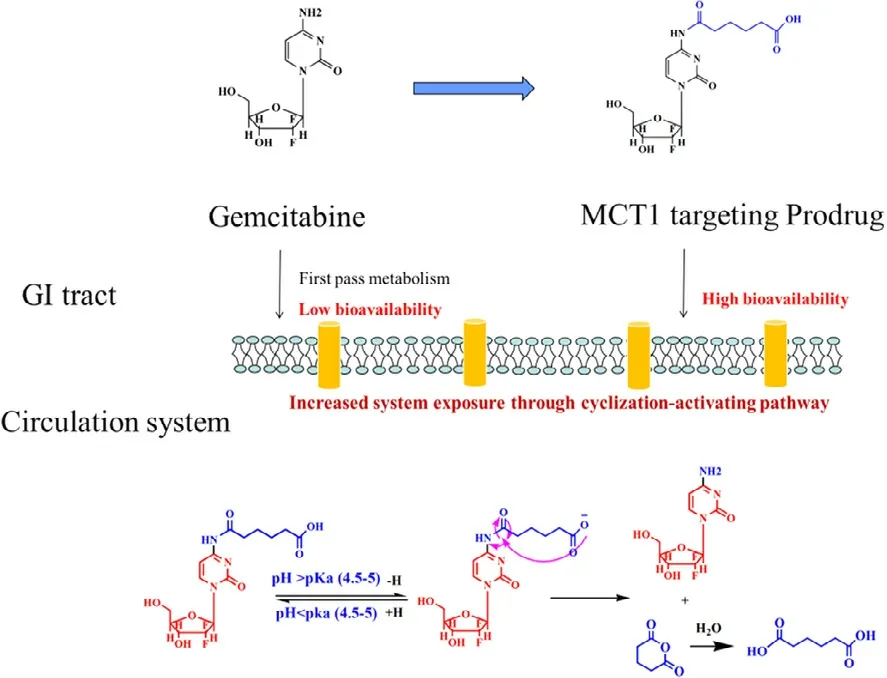
Fig.13–The MCT1-targeting prodrug strategy and the cyclization-activating mechanism of the prodrugs.MCT1:mono-carboxylate transporter protein 1.
The widely expressed intestinal transporters afford new possibilities for oral nanoparticle design.Among them,PEPT1 and OCTN2 are two ideal targets for delivering antitumor drugs [32,34].By contrast,the role of MCT1 as a target to facilitate nanoparticle internalization has been studied rarely until now,although it shows better transport capacity in small molecular prodrugs.Wu et al.linked short chain fatty acids butyrate on classical“mucus-inert”polyethylene glycol(PEG) nanoparticle loading insulin to overcome the mucus barrier,the acidic and enzymatic environment in GI tract.Finally,diabetic modified nanoparticle generated 2.87-fold higher oral bioavailability compared with bare PEG NPs in rats.Moreover,various MCT1 inhibitors,butyrate,proionic acid,lactic acid and pravastatin,significantly decreased the cellular uptake of diabetic modified nanoparticle in Caco-2 cells,indicating the MCT1-mediated endocytosis of nanoparticles [128].In addition,we coupled di-carboxylic acids to the lipophilic polyoxyethylene stearate to synthesize MCT1-targeted polymers loading curcumin.This research is in progress,and we have achieved some preliminary results.Three curcumin-loaded targeted nanoparticles with different chain lengths of di-carboxylic acids as ligands were prepared by emulsion-solvent evaporation,with high encapsulation efficiency and drug loading.Then,the cellular uptake mechanism,endocytosis mechanism and permeability were evaluated to determine the role of MCT1-targeted nanoparticles.In the cellular uptake study,we found that the targeted nanoparticles showed significantly increased intracellular accumulation of coumarin 6 than those of solution in transfected hPepT1-hela cells than mock cells,indicating that MCT1 protein is involved in the enhanced cellular uptake of targeted nanoparticles.The nanoparticle using octanic acid as a ligand exhibited 2.2-to 3.1-fold increased cellular uptake than other nanoparticles,likely due to the improved interaction of the octanic acid linkage with the binding domain of MCT1.Then,the role of co-factor H+on the interaction of nanoparticles to MCT1 was evaluated.The uptake of targeted nanoparticles was increased by 1.9-fold to 2.2-fold when the extracellular pH changed from 7.4 to 6.0,revealing the proton-dependent interaction of the nanoparticle with MCT1 protein.Moreover,the uptake of targeted nanoparticles was significantly decreased at 4 °C than 37 °C,while non-targeted nanoparticles were not affected.Accordingly,the interaction between nanoparticles and MCT1 protein was H+-and temperature-dependent.However,the size of the nanoparticle is too large to enter the cell directly through the channel of the MCT1 protein;thus,the endocytosis mechanism of the nanoparticle should be further evaluated by competitive inhibition experiments using various protein inhibitors.We found that the uptake of nanoparticles decreased in the presence of clathrin and caveolae protein inhibitors,suggesting that the nanoparticles were anchored to the cell membrane through MCT1 and then entered the cell through endocytosis-mediated by clathrin and caveolae proteins.To evaluate the permeability of targeted nanoparticles,insitusmall intestinal perfusion was performed,and theKaandPapp were determined.The targeted nanoparticles both had better absorption than curcumin and non-targeted nanoparticles.Compared to non-targeted nanoparticles,the Kaof octanic acid nanoparticles increased by 1.6–2.9-fold,and thePappincreased by 2.2–3.6-fold in the duodenum,jejunum,ileum,colon and rectum.Additionally,all the targeted nanoparticles showed the highest permeability in the rectum,consistent with the report that the main transport site of MCT1 is the large intestine.Thus,the targeted nanoparticles could enhance the permeability of curcumin due to MCT1.The results of the regulatory effect of the nanoparticles on MCT1,bio-distribution,pharmacokinetics,pharmacodynamics and safety have not been evaluated yet.The above research preliminarily shows that MCT1 can be used as a target to develop nanoparticles.
Pluronic-85,a tri-block copolymer with the form of PEPPPO-PEO,consists of a hydrophilic chain of polypropylene oxide (PPO) and a hydrophobic chain of polyethylene oxide(PEO) [129].Pluronic-85 is used in tablets,suppositories,emulsions and gels and is also used in micelles,resulting in increased solubility,metabolic stability and permeability for drugs [130].In addition,pluronic-85 can be used in combination with an antitumor drug to overcome multidrug resistance due to the inhibition of p-glycoprotein (P-gp) and multidrug-resistant (MDR) proteins [130].Recent research showed that poloxamer-85 is also a substrate of MCT1 protein.In cellular studies,the authors found that the uptake of14C-lactate in bovine brain micro-vessel endothelial cells was abolished by poloxamer-85 at a dose of 0.01% wt.Meanwhile,the lactate levels in the extracellular medium were significantly increased in the presence of various concentrations of poloxamer-85 [131].Furthermore,safety studies indicated no toxicity of pluronic-85 in the liver,kidney,or brain of mice.Therefore,poloxamer-85 can interact with MCT1 protein and can be used as a large molecular ligand for developing oral nanoparticles to increase the oral bioavailability of drugs with poor membrane permeability.However,no relevant studies have been reported.
4.Combination of intestinal OCTN2 and MCT1 for developing oral prodrugs or nanoparticles
The intestinal transporter-targeted strategy has opened a new method of developing oral prodrugs or nanoparticles.For drugs with low membrane permeability,ligands are introduced to parent drugs or carriers to increase the active transport capacity mediated by the intestinal transporter.Several studies have shown that intestinal OCTN2 protein and MCT1 protein are ideal targets for designing prodrugs or nanoparticles.However,a single transporter is easily saturated,thus limiting the transport level of drugs or nanoparticles and resulting in blood concentrations outside the treatment window.It is well known that multiple transporters are expressed in the intestine,and their distribution and function are different.Ininsitusingle-pass perfusion studies,we found that the transport activity of OCTN2 was higher in the duodenum and jejunum,while that of MCT1 was higher in the colon and rectum,although both proteins were distributed throughout the intestine.If the ligands of two transporters are introduced simultaneously to parent drugs or carriers to synthesize double ligandtargeted prodrugs or nanoparticles,the oral bioavailability will be improved due to prolonged intestinal uptake time and increased uptake sites.Compared with the existing technology,a double ligand-targeted strategy has evident advantages:(i) it can achieve higher intestinal transport performance by efficient cooperation of double transporters;(ii) it is a versatile transport system to simultaneously solve the problems of poor permeability,low stability and strong toxicity of drugs;(iii) the system is stable and without immunogenicity.Based on the value of a double ligandtargeted strategy,we are carrying out relevant research on the development of oral prodrugs and nanoparticles.
5.Conclusion
Many factors limit the oral absorption of drugs,resulting in drug candidates that fail in preclinical and clinical trials.Transporters are expressed on intestinal epithelial cells and play a key role in the intestinal absorption of nutrients,thus providing an effective target for the design of oral prodrugs or nanoparticles.In this paper,we focus on intestinal OCTN2 and MCT1 transporters,summarizing the recent advances in transporter-targeting oral drug delivery.Transporter-targeting strategies have been proven successful by simultaneously modifying the permeability,stability and solubility of parent drugs,resulting in higher oral bioavailability.
Despite the rapid development of intestinal transportertargeting strategies,some issues deserve further consideration.These include:(i) the expression of transporters is relatively ubiquitous,and how to decrease the non-targeting distribution of prodrugs or nanoparticles remains a problem;(ii) treatment of diseases often requires a combination of several different drugs,and how to solve the drug-drug interaction mediated by transporters is a big challenge;(iii) there are no crystal structures of transporters,and how to more accurately design transporter-targeting prodrugs remains a bottleneck.Continuous exploration of the underlying mechanisms will help us rationally design improved transporter-targeting strategies.
Conflicts of interest
The authors declare that there is no conflict of interest.
Acknowledgments
This work was financially supported by the Natural Science Foundation of Guangxi Province (Nos.2018JJB140325,2018JJB140377),Guangxi Scientific and Technology Base and Talents of Project (Nos.2018AD19035),Talents Project for Cultivating High-level Talent Teams in the Qi Huang Project of Guangxi University of Chinese Medicine (2018002),the specific subject of the dominant discipline construction of Chinese Pharmacy of Guangxi University of Chinese Medicine,Guang Xi Key Laboratory of Translational Medicine for Treating High-incidence Infectious Diseases with Integrative Medicine and School research projects (no.B170021,2018MS003),and Scientific Research Projects of Guangxi University of Chinese Medicine (B170021,2018MS003).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.ajps.2020.02.002 .
 Asian Journal of Pharmacentical Sciences2020年2期
Asian Journal of Pharmacentical Sciences2020年2期
- Asian Journal of Pharmacentical Sciences的其它文章
- The solute carrier transporters and the brain:Physiological and pharmacological implications
- The role of transporters in cancer redox homeostasis and cross-talk with nanomedicines
- Pharmacologic inducers of the uric acid exporter ABCG2 as potential drugs for treatment of gouty arthritis
- Stimulatory effect on the transport mediated by organic anion transporting polypeptide 2B1
- Amino acid transporters:Emerging roles in drug delivery for tumor-targeting therapy
- Glutamine transporters as pharmacological targets:From function to drug design