
免疫细胞在脓毒血症中的作用及机制研究进展
2020-05-10 03:58:24杨雪李智周宗贞杨勇
药学进展 2020年3期
杨雪,李智,周宗贞,杨勇*
(1. 中国药科大学药物科学研究院,江苏 南京 211100;2. 海南中和药业股份有限公司,海南 海口 570208)
脓毒血症的自然病程分为2 个阶段,早期阶段以促炎性反应为特征,转而进入漫长的免疫抑制阶段。仅有大约10%的患者死于早期的过度炎症反应和难以控制的感染,绝大多数的患者可以渡过早期的急性期,进入相对较长的抗炎和免疫抑制期,而这种状态的持续使原发感染难以控制,同时引起二次感染和医院获得性感染,导致高达90%的患者死于免疫抑制时期,死因是由于免疫细胞功能受损和组织灌流不足而共同导致的多器官功能衰竭。患者在度过最初强烈的促炎反应阶段后,免疫细胞的功能开始受到损伤,包括CD4+和CD8+T 细胞、B 淋巴细胞和树突状细胞(DC)等在各个器官中发生大量凋亡,重要脏器中关键免疫效应细胞的严重凋亡导致了机体的免疫抑制。研究表明防止淋巴细胞凋亡可以提高脓毒血症患者的生存率[1]。凋亡的危害不仅与免疫细胞的严重丢失有关,且与凋亡细胞摄取对存活免疫细胞的影响有关。单核细胞、巨噬细胞和DC 摄取凋亡细胞,通过诱导无功能的细胞增多,促进白细胞介素-10(IL-10)的产生,导致免疫耐受[2]。因此,脓毒血症诱导的细胞凋亡具有多重深刻的影响,其损害了宿主的防御能力。而脓毒血症患者免疫系统发生多种变化,并不是所有的免疫细胞都发生严重凋亡,如嗜中性粒细胞反而凋亡延迟。可见,免疫细胞在脓毒血症期间的免疫应答和维持机体免疫平衡中起关键作用,理解不同免疫细胞群体在脓毒血症中的作用变化和机制或可为脓毒血症的治疗带来新的思路。
1 骨髓来源的抑制细胞与脓毒血症
骨髓来源的抑制细胞(MDSC)是一类髓系来源、未分化完全、具有免疫抑制功能的异质性细胞群体,其包括DC、巨噬细胞、粒细胞的前体等。在正常情况下,未成熟的骨髓细胞(IMC)可以分化为成熟的免疫细胞。然而,在某些病理条件下,这种终末分化常常被抑制,导致异质性群体IMC 的积累,即MDSC[3]。这种异质群体不能分化成正常的免疫细胞,体外培养晚期的MDSC,发现其缺乏离体分化的能力[4]。根据表面标志物及细胞核的形状可以将小鼠MDSC 分为粒细胞型和单核细胞型[5]。在小鼠脓毒血症的盲肠结扎穿孔(CLP)模型中,随着病程的发生,MDSC 在骨髓中的比例显著扩大,晚期MDSC 高达骨髓细胞的88%[4],在严重脓毒血症或脓毒血症感染性休克的患者中也同样检测到MDSC 数目的增加[6]。
MDSC 在各种炎症和感染的刺激条件下生成,在脓毒血症中最重要的特点是其免疫抑制功能,MDSC 通过产生免疫抑制物质和抑制T 细胞的增殖和活化来减弱先天性和获得性免疫反应[7]。脓毒血症早期小鼠的MDSC 产生高水平的促炎性细胞因子:肿瘤坏死因子-α(TNF-α)和IL-6 以及一氧化氮(NO),而晚期脓毒血症小鼠产生更高水平的抗炎性细胞因子IL-10、转化生长因子-β(TGF-β)和精氨酸酶(Arg)[5]。MDSC 的另外一个重要的免疫抑制功能是抑制T 细胞活化、功能和增殖能力,MDSC 能够显著降低CD4+T 细胞的增殖以及干扰素-γ(IFN-γ)的分泌,并且不同种类的MDSC 调节T 淋巴细胞的机制不尽相同,其中单核细胞型MDSC 调节活性氧类(ROS)的含量,通过修改T细胞抗原受体-ζ 链(TCR-ζ 链)干扰T 细胞发挥功能,而粒细胞型则增加NO 的含量及Arg 的活性,破坏下游IL-2 受体的信号传导途径,促进T 细胞凋亡[8]。
许多因素能加速脓毒血症过程中MDSC 数量的扩大。例如:①粒细胞集落刺激因子(G-CSF)、巨噬细胞集落刺激因子(M-CSF)、IL-6 以及血管内皮生长因子(VEGF)与骨髓造血干细胞表面相应的受体结合后,激活细胞内Janus 激酶/信号转导和转录激活因子3(JAK/STAT3)信号通路,导致还原型辅酶Ⅱ氧化酶(Nox2)、活化转录因子CCAAT 增强子结合蛋白β(C/EBPβ)的表达增加,从而诱导MDSC 的增殖[9-10];②IL-4 和IL-13与MDSC 细胞表面受体结合,通过激活细胞内的信号转导和转录激活因子6(STST6)信号通路来诱导MDSC 形成[11];③细菌脂多糖(LPS)、热休克蛋白72(HSP72)及S100A8/A9 蛋白可通过激活MDSC 表面的Toll 样受体(TLR)继而激活髓样分化基因MyD88,最后通过核转录因子(NF-κB)来诱导MDSC 形成[12]。
2 嗜中性粒细胞与脓毒血症
嗜中性粒细胞数量约占血液循环中白细胞的70%,在骨髓中分化发育后,进入血液或组织来发挥作用。其主要作用是释放、迁移和吞噬。在脓毒血症早期,较多未成熟嗜中性粒细胞从骨髓储存库中释放出来以补充循环粒细胞,随后嗜中性粒细胞被招募并迁移至外周组织。大多数嗜中性粒细胞在骨髓中释放后24 h 内发生细胞凋亡,而在脓毒血症中,与细胞凋亡加速的其他淋巴细胞相反,中性粒细胞的凋亡发生延迟[13]。由于未成熟的嗜中性粒细胞释放增加和循环中性粒细胞凋亡的延迟,败血症患者体内通常可见不同程度的成熟循环中性粒细胞数量的显著增加[13]。
从严重脓毒血症患者体内分离出的嗜中性粒细胞中发现,起抗凋亡关键作用的Mcl-1 蛋白水平增加[14]。另外,LPS 和外周循环中的补体成分5a(C5a)可诱导细胞外调节蛋白激酶(ERK1/2)和嗜中性粒细胞中的磷脂酰肌醇-3-羟激酶(PI3K)的活化,导致抗凋亡蛋白Bcl-xL 的表达增加和Bim 表达水平降低。磷酸化的Akt 也可导致Bad 的磷酸化,从而阻止凋亡体的形成并抑制嗜中性粒细胞凋亡[15]。此外,暴露于LPS 会降低酪氨酸磷酸酶-1(SHP-1)的活性及其Src 同源结构域2 与Caspase-8 的结合作用,伴随着非受体酪氨酸激酶在嗜中性粒细胞中的表达增加,从而降低嗜中性粒细胞自发凋亡的速率[16],并且,Caspase-8 的活性降低导致髓样细胞核分化抗原(MNDA)的切割或重定位受损,同时Mcl-1 积累,这几个因素一起导致嗜中性粒细胞凋亡的延迟[17]。其他细胞凋亡途径如TNF-α 或Fas 配体(FasL)介导的外源性途径,以及嗜中性粒细胞上的一些特异性受体也被认为可能导致脓毒血症嗜中性粒细胞凋亡的延迟[18]。
免疫抑制阶段的脓毒血症动物模型和患者中,嗜中性粒细胞的功能都有明显改变,包括细菌清除受损、反应性降低、产生ROS 物质,以及招募至感染组织的嗜中性粒细胞数量明显减少等[19]。嗜中性粒细胞迁移的损害与脓毒血症的预后不良密切相关,可能有以下几种原因:①嗜中性粒细胞在趋化因子配体8(CXCL8)的作用下黏附于内皮细胞,CXCL8 与高亲和力受体CXCR1 和CXCR2 结合,通过激活PI3K 和张力蛋白同源途径促进嗜中性粒细胞的趋化。与健康人相比,脓毒血症患者的嗜中性粒细胞表面趋化因子受体CXCR2 和β 整合素CD11b 的表达减少[20],嗜中性粒细胞对IL-8 的趋化性也降低。另有研究表明,严重的脓毒血症中,嗜中性粒细胞中的TLR9 激活,触发G 蛋白偶联受体激酶2 表达和CXCR2 下调[21]。②脓毒血症炎性细胞因子和LPS 介导的诱导型一氧化氮合酶(iNOS)上调,导致NO 大量增加,从而减弱中性粒细胞的滚动和黏附。另外,NO 还参与TNF-α、IL-8 以及巨噬细胞介导的嗜中性粒细胞趋化失调[21]。
3 树突状细胞与脓毒血症
DC 在脓毒血症中表现出明显凋亡。在对脓毒血症患者的尸检报告中发现循环和脾脏DC 的数量以及被DC 占据的脾脏面积百分比显著减少[22]。而DC的损失在脓毒血症死亡患者中比存活者更严重,并且脓毒血症患者后期不仅DC 数量减少,存活的DC 抗原提呈能力降低,表达人类淋巴细胞抗原-D受体(HLA-DR)水平也降低,IL-10 分泌却增加[23]。此外,脓毒血症患者单核细胞来源的DC 不能诱导强有力的效应T 细胞反应,而是诱导T 细胞失活或调节性T 细胞(Treg)增殖[24]。在小鼠烧伤模型中,DC具有免疫抑制特性,对细菌感染的防御能力减弱。已有报道,预防脓毒血症诱导的DC 凋亡或改善DC功能,可提高患者生存率[25]。在DC 中选择性过表达抗凋亡因子Bcl-2 的小鼠在LPS 休克模型中存活率明显提高[26],这些过表达Bcl-2 的DC 对脓毒血症诱导的凋亡具有抵抗作用,表明DC 的死亡是决定脓毒血症存活率的一个重要因素。
FMS 样酪氨酸激酶3 配体(FLT3L)是一种DC 生长因子,可诱导DC 数量快速增加。当小鼠受到铜绿假单胞菌感染时,用FLT3L 治疗烧伤小鼠可以恢复DC 功能并改善小鼠存活率。并且,经FLT3L 治疗的小鼠其DC 分泌IL-12、IL-15 和IFN-γ的水平增加,对CD4+T 细胞、自然杀伤细胞(NK)和中性粒细胞的功能也有广泛影响[25]。肺内转移骨髓源性的DC,能使DC 发挥正常的功能,从而预防致死性烟曲霉感染的小鼠死于原发性腹膜炎[27]。TLR 激动剂也能降低出血性休克后小鼠的肺炎死亡率,其作用机制可能是TLR 激动剂使DC 上主要组织相容性复合体Ⅱ(MHC II)类分子、CD80 和CD8 表达增加[27],因此,一些研究者认为保护和恢复DC 功能应是脓毒血症治疗的一种重要策略。
4 单核细胞和巨噬细胞与脓毒血症
在脓毒血症的最初阶段,巨噬细胞表面的模式识别受体识别并结合病原体表达的病原体相关分子模式(PAMP),激活白细胞,释放大量炎症因子,发挥促炎作用,促进宿主防御,而后期,在脓毒血症患者中LPS、其他TLR 激动剂和各种其他细菌化合物的作用下,单核细胞释放促炎细胞因子的能力逐渐下降,这一发现与LPS 耐受中的现象一致。脓毒血症患者的单核细胞分泌TNF、IL-1α、IL-6和IL-12 等减少,而IL-10、TGF-β 等抗炎介质的释放能力不受损或增强[28]。这些发现表明LPS 仍可激活单核细胞,但细胞内信号转导偏向于产生抗炎相关分子的方向,这符合了单核细胞重编程的概念[29]。
LPS 耐受的机制至今还不完全清楚。mRNA 表达水平分析显示,编码抑制性信号分子和抑制性细胞因子的基因表达增加,促炎因子和趋化因子受体基因表达减少[21]。最近的研究强调了表观遗传调控的重要作用,如组蛋白修饰和染色质重构。在体外,这些表观遗传修饰恢复了IFN-γ 的激活和单核细胞细胞因子的释放[30],因此,IFN-γ 可能通过促进TLR 诱导的染色质抵抗LPS 耐受。
LPS 耐受对单核细胞和巨噬细胞产生的2 个主要结果是免疫抑制介质(主要是IL-10)的释放增加和通过减少HLA-DR 表达导致抗原递呈减少,这两者都与脓毒血症较差的预后有关。IL-10 的持续释放可能导致或增强脓毒血症诱发的免疫抑制,从而增加继发性微生物感染的易感性[31]。在临床相关的脓毒血症动物模型中,阻断IL-10 可以逆转动物体内的LPS 耐受及脓毒血症诱发的免疫抑制,提高生存率[32]。
单核细胞HLA-DR 的低表达可作为单核细胞失活的标志[33]。已报道,低表达的HLA-DR 与单核细胞功能受损密切相关,例如,由于抗原提呈功能受损,细菌感染后TNF 和IL-1β 的释放水平降低,破伤风毒素感染后淋巴细胞增殖减少[34]。单核细胞HLA-DR 表达降低还与医院感染和死亡风险增加有关。单核细胞HLA-DR 的表达下降是脓毒血症发生率和死亡率的独立预测因子。这一发现证明单核细胞失活和免疫抑制都增加了脓毒血症的不良事件风险。
5 自然杀伤细胞与脓毒血症
NK 在外周血中的数量较低,尤其是在脓毒血症中,这使得对该细胞亚群的研究变得困难。在脓毒血症患者的免疫抑制阶段,循环NK 数量明显减少,常持续数周,并与患者死亡率增加有关。在动物模型、脓毒血症患者、烧伤和创伤性损伤患者中NK 的细胞毒功能减弱,细胞因子分泌减少[35]。
在多种细菌感染的脓毒血症中,NK 对于TLR激动剂的细胞因子产生反应减弱,表明NK 对TLR激动剂产生了耐受性,从而再现了单核细胞LPS耐受性的一些特征[36]。NK 产生IFN-γ 的功能受损也同样出现在脓毒血症患者中。由于NK 在抗病毒防御中起重要作用,可以推测,NK 功能受损有利于潜伏病毒的重新激活[37]。脓毒血症患者中NK产生IFN-γ 的功能受损可能是导致继发感染率上升的一个重要因素,也是单核细胞HLA-DR 表达减少的原因[38]。
6 CD4+辅助性T 细胞与脓毒血症
许多研究报道了脓毒血症对循环和组织T 细胞的深远影响,其中一些最显著的影响发生在CD4+T细胞上。根据刺激产生的细胞因子类型,成熟CD4+Th 细胞已被鉴定为Th1、Th2 和Th17 细胞亚群。早期研究表明,Th1 和Th2 细胞相关细胞因子的产生均在脓毒血症和创伤的初始免疫反应中降低[39]。进一步的研究显示,T-bet 和GATA3 表达显著降低,这2 种转录因子分别调节Th1 和Th2 细胞的反应,强化了Th1 和Th2 细胞在创伤和脓毒血症中被抑制的观点[40]。与T-bet 和GATA3 不同,Treg 相关转录因子FOXP3 的表达没有发生改变。组蛋白甲基化和染色质重塑可通过作用在IFNG和GATA3基因的启动子区域抑制Th1、Th2 细胞的功能[41]。
虽然Th1 和Th2 细胞亚群的研究较少,但目前普遍认为Th17 细胞通过产生IL-17 和IL-22 在保护细胞抵抗细菌和真菌感染方面发挥重要作用[42]。脓毒血症中Th17 细胞反应减弱,可能是由于Th17细胞特异性转录因子视黄酸受体相关孤儿受体γt(RORγt)表达减少[40]。脓毒血症患者中Th17 细胞表型的这种缺陷可能是导致继发性真菌感染易感性增加的一个因素。研究证明,IL-7 治疗可增加Th17细胞反应,减少继发性白色念珠菌感染的死亡率[43]。
7 调节性T 细胞与脓毒血症
在脓毒血症患者和动物模型中,血液或脾脏中Treg 的比例随着病程显著增加,但是这种相对增加是由于效应T 细胞数量的减少,而不是Treg 绝对数量的变化[44]。这一发现提示Treg 对脓毒血症诱导的凋亡更有抵抗力,可能是由于抗凋亡蛋白Bcl-2 表达增加所致。热休克蛋白和组蛋白在脓毒血症中表达增加,它们作为Treg 的强诱导剂,在脓毒血症中也促进Treg 数量的增加[45]。
在脓毒血症中,Treg 增多不仅与较差的预后相关,还与效应T 细胞增殖和功能下降有关。这些抑制作用可通过使用Foxp3 特异性的小干扰RNA (siRNA)阻断Treg 分化而被完全消除。此外,脓毒血症诱导的免疫抑制可通过Treg 促进实体肿瘤快速生长[46]。Treg 也可以抑制先天性免疫细胞。在LPS刺激下,Treg抑制单核细胞和中性粒细胞功能[47]。总之,有大量证据表明,脓毒血症和创伤患者的Treg 数量增加,这些细胞通过作用于先天免疫细胞和适应性免疫细胞,损害机体免疫功能,并导致患者死亡。
8 免疫细胞间的相互作用
脓毒血症发作后,机体的促炎免疫反应迅速激活,先天免疫系统细胞释放大量的促炎细胞因子,以杀灭病原体,促进宿主防御。最初的炎症反应的方向、强度和持续时间由宿主因素和病原体因素共同决定。如果持续下去,机体的先天和适应性免疫系统发生关键变化,进入一个漫长的免疫抑制状态,患者将可能死于不能明确的二次感染。研究证明,在脓毒血症小鼠中,由巨噬细胞产生的高水平TGF-β 以旁分泌方式作用于T 细胞表面受体,促进第二信使环磷酸腺苷(cAMP)产生,从而使T 细胞释放IL-2 的能力下降,抑制其增殖能力,TGF-β 还可促进T 细胞分泌IL-10,进一步抑制T 细胞增殖[48]。同时,脓毒血症小鼠的DC 在诱导T 细胞增殖方面比对照组小鼠的DC 更弱,在DC 和T 细胞的共培养体系中,对照组小鼠T 细胞分泌的IL-2 水平显著低于脓毒血症小鼠T 细胞,DC 分泌的抑制性细胞因子IL-10和IL-4严重限制了免疫细胞活化和扩张的强度,促进大量Treg 产生并负调控炎症反应,细胞外的高迁移率基团蛋白(HMGB1)也进一步介导DC 促进Treg 的免疫抑制功能,并调节巨噬细胞和T 淋巴细胞的功能[2]。此外,巨噬细胞通过前列腺素E2(PGE2)和吲哚-2,3-双加氧酶(IDO)刺激IL-10 释放增加,PGE2 和IDO 也抑制DC 的分化、成熟和抗原表达过程,与间充质干细胞(MSC)产生的人白细胞抗原G5(HLA-G5)和IL-10 发挥共刺激作用时,这些PGE2 和IDO 也能抑制NK 的活化和增殖,且HLA-G5 以与IDO 和PGE2 相似的作用机制对T 淋巴细胞增殖产生影响[49]。在脓毒血症患者的病程后期,大量骨髓来源的DC、单核巨噬细胞以及嗜中性粒细胞表型功能发生转变,这部分细胞源源不断地补充MDSC,发挥了强烈的免疫抑制功能,这也揭示了免疫细胞发挥功能的过程从来就不是孤立存在的。脓毒血症免疫抑制时期各种免疫细胞间的相互作用使免疫系统构成一个复杂的调控网络(见图1),了解其整体变化对于脓毒血症的治疗来说至关重要。
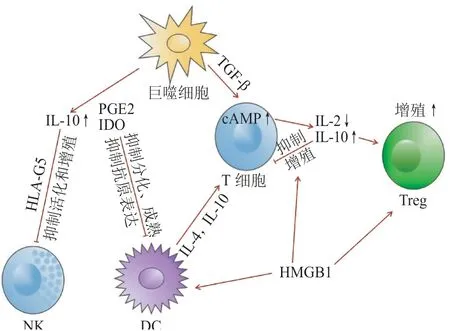
图 1 脓毒血症的免疫抑制时期各种免疫细胞之间的相互作用Figure 1 Interaction of immune cells during immunosuppression of sepsis
9 结语与展望
在急诊室和重症监护室中,严重脓毒血症和感染性休克发生率高并普遍存在,导致患者先天性和适应性免疫反应广泛激活和功能障碍。脓毒血症中一些主要的免疫细胞(包括骨髓来源的抑制细胞、嗜中性粒细胞、巨噬细胞、DC、淋巴细胞、NK)通过调节多种受体表达或细胞因子分泌对脓毒血症过程中免疫反应性的调节发挥了重要作用,从而影响脓毒血症的病情发展和结果。尽管医疗技术快速发展,护理方法广泛改善,更多抗菌药物可供选择,这些治疗策略未能充分降低严重脓毒血症患者的死亡率。已有很多报道证明免疫细胞在脓毒血症先天和后天免疫反应中的关键作用及适应性变化,如骨髓来源的抑制性细胞数量大量增加,中性粒细胞凋亡延迟等[3,13]。近年来,多种针对脓毒血症中免疫细胞异常的药物也被应用于临床,如胸腺五肽能提高ICU 内脓毒血症患者的CD3+、CD4+细胞水平及CD4+/CD8+比值[50],胸腺肽α1 也能明显提高CD4+/CD8+比值,促炎细胞因子如TNF、IL-6,抗炎细胞因子包括IL-4、IL-10 快速达到平衡。这些药物可明显恢复和提高脓毒血症患者的免疫功能[51]。因此,了解脓毒血症模型动物及患者体内免疫细胞的变化,有利于正确评估患者的免疫状态并为患者制定新型免疫调节策略,从而改善脓毒血症患者生存率及预后,对脓毒血症的治疗具有重要的临床意义。
猜你喜欢
现代畜牧科技(2021年7期)2021-07-28 06:41:00
兽医导刊(2019年1期)2019-02-21 01:13:54
兽医导刊(2016年12期)2016-05-17 03:51:17
中国药理学与毒理学杂志(2015年3期)2015-12-16 09:11:40
温州医科大学学报(2014年7期)2014-07-18 02:43:14
云南畜牧兽医(2014年5期)2014-02-28 21:25:39
当代畜禽养殖业(2014年10期)2014-02-27 07:59:49
现代检验医学杂志(2014年6期)2014-02-02 03:02:25
现代检验医学杂志(2014年5期)2014-02-02 02:51:34
食品科学(2013年15期)2013-03-11 18:25:40
