
单层MoS2在合金化及应力调控下光催化裂解水产氢的理论研究
2020-05-06 03:27徐祥福赖国霞李天乐许诗圳陈星源朱伟玲
燃料化学学报 2020年3期
徐祥福, 陈 佳, 赖国霞, 李天乐, 许诗圳, 陈星源, 朱伟玲
(广东石油化工学院, 广东 茂名 525000)
氢能作为一种清洁和可持续能源,被视为是化石能源替代者之一。其中,利用可见光催化裂解水产氢(HER)是绿色环保的方式之一,吸引了很多研究者关注[1]。直接分解水需要很大的能量,需借助催化剂进行。可见光分解水制氢分为半反应和全反应制氢,全反应制氢即不需要牺牲剂直接利用太阳光分解水,并且是一个可持续进行的反应,这就要求催化剂能带的CBM和VBM跨越标准H+/H2和O2/H2O电位。为了吸收占太阳光能量最大部分的可见光,还要求催化剂的带隙为2 eV左右,最大不能超过3 eV[2],为了使全反应制氢能够持续进行,还要求CBM与H+/H2电位差和VBM与O2/H2O电位差尽可能相等,保持热力学的平衡[3]。总之,对裂解水产氢催化剂来讲,禁带宽度、带边位置是其基本的决定因素。
常见的催化材料除了效率最高的贵金属以外,还有低成本的d0、d10过渡金属离子的氧化物、氮化物、硫化物[4]。除此之外,近期理论及实验研究揭示二维材料因其独特的电子性质、力学性质、光学性质[5]表现,特别是边缘催化活性位置[6]的暴露,明显提高了催化效率。二维材料区别于体材料在催化方面主要有两点优势,首先,二维材料其比表面积较大,比较适合光吸收,同时也能提供光催化反应足够的场所;第二,二维材料都是原子级的厚度,光致电子和空穴可以快速迁移至水界面进行光催化反应[7],减少载流子复合机率,从而提高催化效率。
二维光催化材料中的MoS2,以及其他硫族化合物的二维形式材料,如GaSe、InTe[8]等,成为近几年研究热点。MoS2体材料是间接带隙,大小为1 eV,随着层数的减少,带隙增大,能带结构由间接带隙转变化直接带隙。单层的二维MoS2为直接带隙,大小为1.75 eV[5,9,10],这与可见光光催化制氢要求的最佳2 eV[1,2]接近,且因是直接带隙,增加了光致电子空穴的产生机率,比较适合可见光光催化制氢,这早已被实验所证实[11]。另外,MoS2二维材料因其网状结构,耐张力性要强于体材料,目前在实验上,利用调节应力来调节电子性质,以提高分解水效率[12-14]也是研究的一个方向。但MoS2在光催化分解水应用上,也有缺点,一是因为其带隙偏低,离最佳2 eV仍有差距;二是其导带边(CBM)位置距离标准氢电极还原电位H+/H2太小,其氧化和还原能力不平衡,用HSE06方法计算出来的其导带边(CBM)在标准氢电极还原电位H+/H2之上0.19 eV,而价带边(VBM valence band maximum)在O2/H2O氧化势之下0.60 eV[7,8,15],导带底和价带顶的电位相对于水的氧化势与还原势很不均衡,最终HER反应会终止。这限制了单层MoS2在可见光下全分解水的应用。所以为改善这两方面缺点,一是增大其带隙;另一个是提高导CBM带边位置。
为了改善单层MoS2的能带结构,提高CBM带边位置,通常的策略是通过掺杂来改变带边位置[9,16],掺杂一般是在禁带中产生杂质能级从而改变带边位置,这样做虽然改变了带边位置,但也成为了载流子复合的中心,催化效率反而会降低。相较之下,采用合金的方法,形成的是能带,既改变了带边位置,也能保证载流子寿命不受太大影响[9,17-19]。据研究报道与MoS2同族元素构成的结构相近的MoSe2、MoTe2、WS2的材料性质与MoS2相近,可应用于光催化分解水[20],所以本实验的研究对象为单层二硫化钼(momolayer-MoS2)与MoSe2、MoTe2、WS2的合金材料。
除合金化手段调控之外,理论和实验中用施加应力的方法调节MoS2电子性质、能带结构也能起到很好的效果[14,13,21]。但据目前掌握的文献,都是采用单一调节手段,极少研究报道采用合金方法之后,再施加应力,在这两种调控手段下,催化剂性质变化情况,能否改善催化效率,值得深入研究。
因此,本研究基于密度泛函理论(DFT),对二维的单层二硫化钼(momolayer-MoS2)进行不同类型的合金化(分别与MoSe2、MoTe2、WS2),且加入应力,研究在这两种调控手段下,其电子性质、能带结构、带边位置和对光催化裂解水产氢的影响。
1 计算方法
1.1 计算模型
本研究的对象是单层MoS2及其合金体系,单层MoS2原胞如图1(a)所示,为半导体态2H结构,拥有三个原子层,两个S原子层把Mo原子层包夹在中间,形成了类似三明治结构的S-Mo-S模型。合金模型如图1(b)、(c)、(d)所示,分别是与MoSe2、MoTe2、WS2形成合金。超胞采用3×3体积建模,浓度为12.5%。为了防止周期性边界条件和层与层之间弱范德华力对单层MoS2的影响,作者沿C轴方向设立了2.5 nm的真空层,优化后的晶格参数见表1。
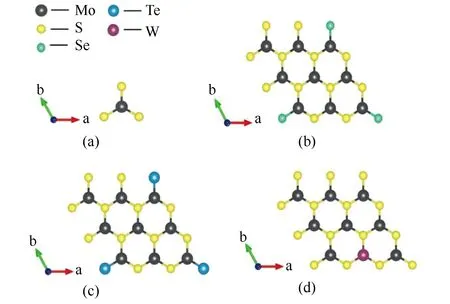
图1 MoS2(a)及其合金(Mo9S17Se(b)、Mo9S17Te(c)、Mo8WS18(d))结构示意图
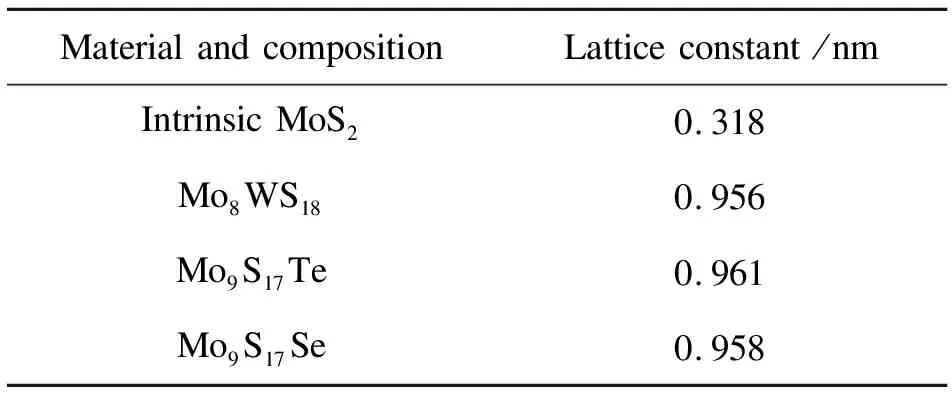
表1 优化后晶格常数及组成比例
1.2 计算方法及过程
本研究计算基于DFT(Density functional theory)的Vienna ab-initio Simulation Package(vasp)软件包,采用GGA交换关联函数,PAW赝势,其平面波的截断能为400 eV,计算时模型真空层为2.5 nm。二维本征MoS2沿a、b轴方向使用Monkhorst-Pack 方法以Gamma点对称取25×25×1 k点,合金化MoS2超胞选取9×9×1 k点。计算过程能量收敛标准为10-8eV,力的收敛标准为0.1 eV/nm,所有的结构都是经过充分弛豫的。据Li[22]报道,MoS2的拉力在施加2%力时,仍符合线性杨氏模量方程,当超过2%后,能带结构会逐渐由半导体属性转变为金属属性。所以本研究选择施加2%双轴拉应力和压应力。
2 结果与讨论
2.1 结构分析
经充分弛豫、合金化前后的键长、键角见表2。通过计算得到本征MoS2的原胞基矢a=0.318 nm,这与实验值0.320 nm[23]接近,并与其他报道一致[24],证明了本计算方法的正确性。由表2可以看出,与WS2合金,其键长变短,键角变大,在c轴方向类似受到压力的作用,推测原因,是由于W原子半径较大,由于MoS2是三原子层结构,Mo处于中间层,受上下两层作用,W替位Mo原子位置后基本没发生变化,所以键长变小;另一方面原因,推测是由于W的电负性比Mo要强,引起键长变小。同理,用Te、Se替代S进行合金,由于Te、Se原子半径较S要大,电负性比S弱,所以键长变长,键角变小,在c轴方向呈现出拉力的效果。结构上的变化肯定会引起能带结构和电子性质的变化。
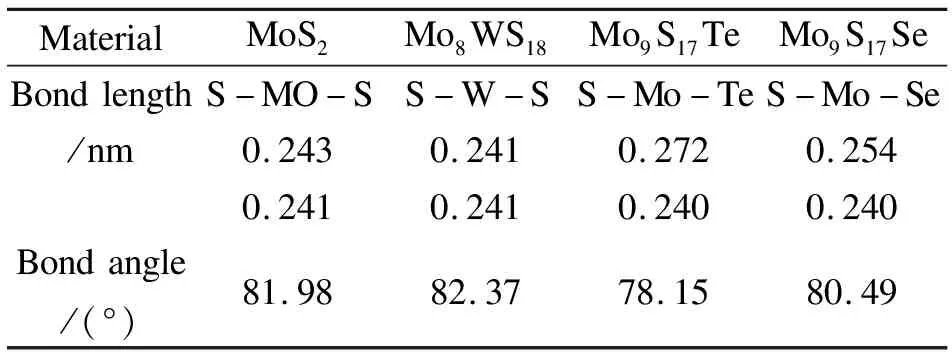
表2 结构优化后的键长参数
2.2 合金化对带边位置的影响
合金化后材料的带边位置如图2所示,所有数值计算都采用标准方法,相对于绝对真空能级[3,8],标准氢电极水的氧化电位取-4.43 eV,还原电位取-5.66 eV,图2(a)为不加应力的带边位置图,由图2(a)可以看出,本征的MoS2氧化与还原能力最不均衡,CBM带边与H+/H2比较接近为0.187 eV,而VBM带边与O2/H2O稍远为0.274 eV。
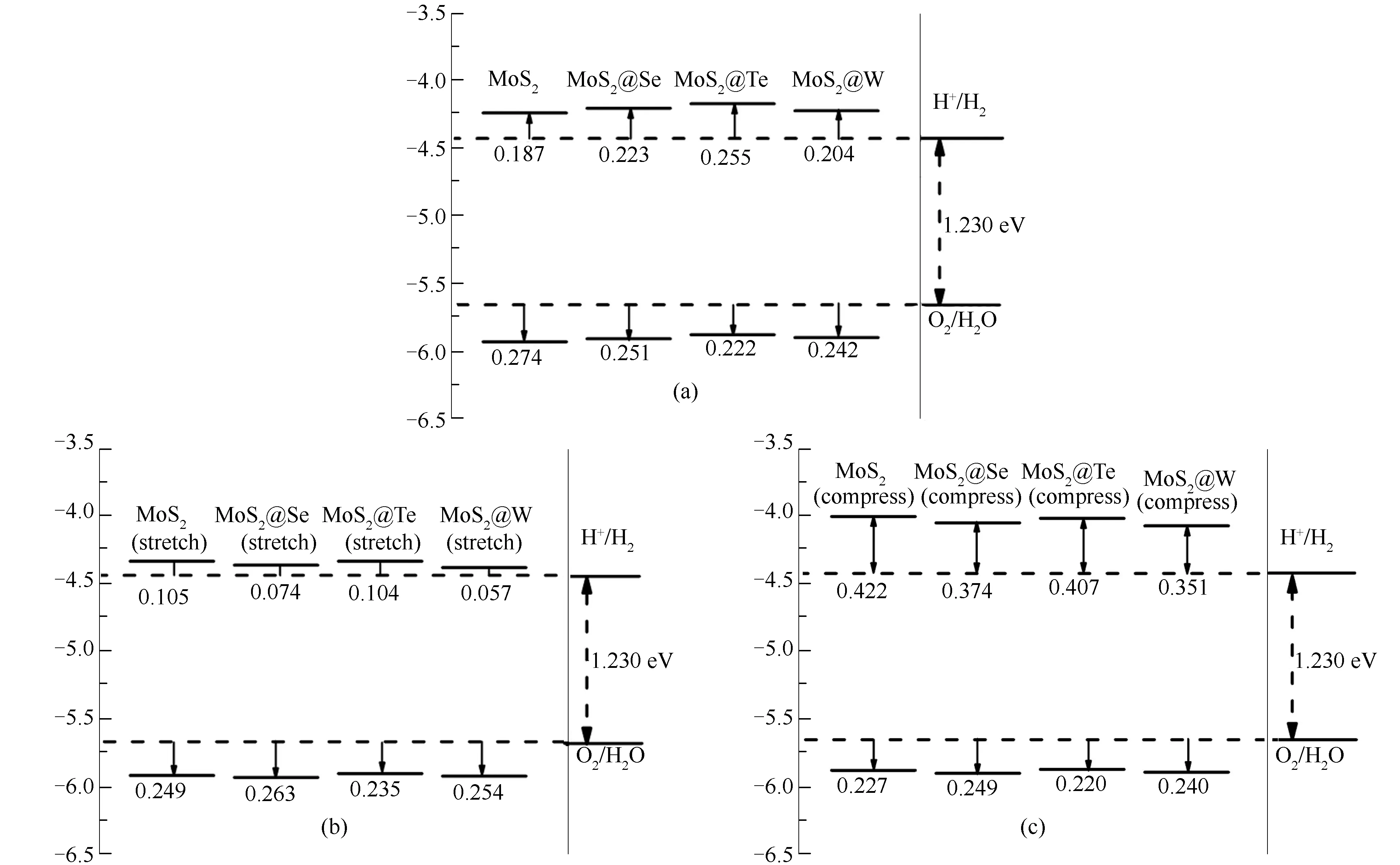
图2 不施加力(a)、施加拉应力(b)和施加压应力(c)的带边位置图
但经过合金化以后,CBM带边位置都有不同程度的上升,尽管VBM也表现出上升趋势,但CBM上升幅度更大,有利提高分解水效率,这与报道[25]中Se对MoS2影响一致。究其原因是由于本征MoSe2、MoTe2、WS2的CBM带边位置比MoS2高0.30-0.40 eV[15],按照与MoS2CBM带边差距对比,排列由大到小依次是MoTe2>MoSe2>WS2,与本研究合金化后的差距大小排序一致。由图2(a)还可知,合金化后,虽然上移了CBM带边位置,但由于本征MoSe2、MoTe2、WS2的VBM价带边位置位于O2/H2O之上,只能进行半分解水反应,合金化后导致小幅度提高了VBM带边位置,使带隙并没有明显增大。总之,经过合金化后,CBM带边位置得到改善,有利于催化效率的提高,但这几种材料带隙仍都为1.7 eV左右,相较2 eV偏小。
加了拉应力的四种材料的带边位置如图2(b)所示,加了拉应力后,带隙明显减小,为1.54-1.58 eV,同时CBM带边位置全部下移,除此之外,VBM带边位置除了本征MoS2之外,也是全部下移。也就是说带隙变小,离裂解水最佳带宽2 eV更差了,而且CBM、VBM带边位置全部下移,加剧了氧化还原势不平衡度,使催化制氢效率降低。所以对合金材料施加拉应力对催化效率是严重损害的。
加了压应力的四种材料的带边位置如图2(c)所示,施加压应力后,带隙明显增大,在1.83-1.88 eV。合金化的三种材料加压应力与拉压应力对带边位置影响不同,施加压应力下,CBM带边位置有明显的上升,位置变化幅度除本征MoS2外相差不大,约为0.15 eV,最重要的是,VBM带边位置几乎无变化,这对于MoS2原本CBM氧化势差距小,VBM还原势大的不均衡性[20]会有较大改善。
施加压应力后,本征的MoS2CBM带边位置变化最大,与标准氧化电位的差距,由不施加应力的0.187 eV,变化到施加压力的0.422 eV,是这几种材料当中,带边位置最高的,也是变化幅度最大的,由此可以看出,本征MoS2对压应力的反馈要比合金更敏感,可以预计如果单独以应力为调控手段调节MoS2带边位置的实验可操作性相较合金材料会差。
由上面分析可知,施加拉压力对于光催化制氢应用并无益处,而施加压应力可以提高CBM带边位置、增大其带隙,可以提高光吸收及平衡氧化还原势,对提高光催化效率是有帮助的,因此,在此基础上,本研究分析了施加压应力和不施加力时MoS2及其合金材料的能带结构。
2.3 能带结构
能带结构是分析半导体光电性质时候的重要依据,合金化将会改变晶体中原子质量比以及它的晶格参数等属性,也导致其能带结构发生变化。
图3为没有施加应力((a)、(b)、(c)、(d))与施加压应力情况((e)、(f)、(g)、(h))下材料的能带结构图。
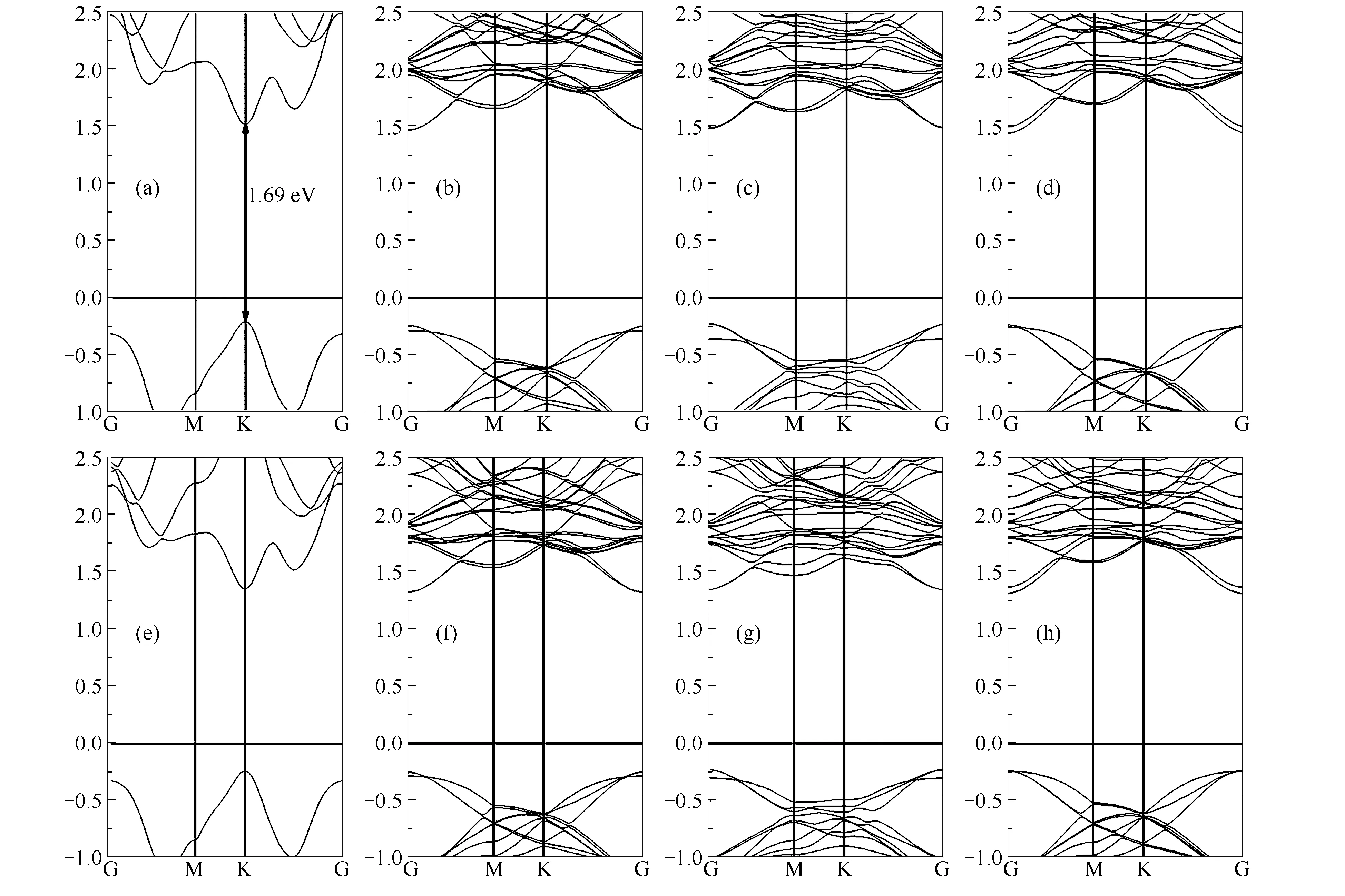
图3 MoS2及其合金的能带结构示意图
由图3可知,没有施加应力时,本征MoS2是直接带隙,其余三种合金材料,由于其结构的对称性改变,变为间接带隙。这三种合金材料的能带结构相似,原因是MoTe2、WS2、MoSe2三种材料的能带结构相似[20],所以合金化后,能带结构仍相似。施加压应力后,可以看到价带顶没有太大变化,而不同材料,导带底在M与K点有明显下降,这与前面带边位置的计算结果相一致。为了更明确产生此现象的原因是否是合金元素的影响,本研究计算了四种材料施加压应力前后的分波态密度(PDOS,partial density of states)以及总态密度(TDOS, total density of states)。
由于半导体材料吸收光产生的电子空穴载流子密度都是在带边位置最大,所以通常在费米能级附近的DOS(density of states)图比较有意义,图4给出了费米能级附近价电子分波态密度图。由图4可知,本征MoS2的CBM与VBM带边位置主要是由Mo 4d轨道贡献,次要是由S 3p轨道贡献,这与文献中描述一致[26]。增加压应力以后,可以看到VBM位置变的陡峭,带隙变大。
进行合金化以后,三种合金材料在VBM位置表现出相似的情况,由图4还可以看出,VBM位置主要贡献仍然是Mo 4d轨道,尽管合金元素也有贡献,但由于原子比例较少,所以贡献较小。这区别于通过掺杂方式进行调节能带结构的情况[2,4],掺杂是由于杂质原子在禁带中产生杂质能级改变带边位置和带隙,杂质能级一般情况下比较局域,而且会成为载流子复合的中心,使载流子寿命降低。而本研究三种合金材料CBM、VBM带边位置,是Mo 4d、S 3p、Te 4p、Se 5p、W 5d轨道整体的贡献,合金元素对带边的贡献不大,这与掺杂方式有本质的区别,合金化材料的电子结构形成的是能带而不是孤立的能级,对载流子寿命影响不大。施加了压应力以后,由图4(e)、(f)、(g)、(h)可以看出,CBM带边位置的DOS变化不大,而VBM带边位置向高能量处移动,移动的方式是整体向右移,这也与能带结构的改变相对应。
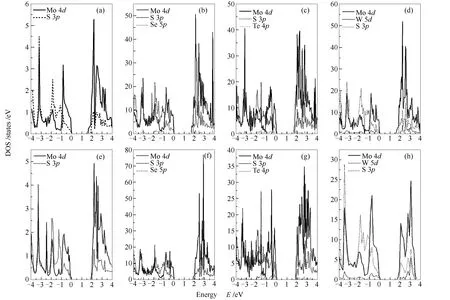
图4 三种合金材料在费米能级附近的PDOS谱图
由此可以看出,通过合金化方式对MoS2材料进行改性,既可以调节带边位置、带隙大小,又不会形成复合中心,施加压应力以后,可以进一步增加带隙,最关键的是在不怎么改变VBM带边位置的情况下,能够提高CBM带边位置,这有利于MoS2催化效率。
3 结 论
本研究在密度泛函理论第一性原理的基础上,计算了二维材料MoS2分别与MoSe2、MoTe2、WS2合金后的电子能带结构、态密度以及带边位置的变化。研究发现,单层MoS2具有直接光催化分解水的能力,但因其导带底电位相对于水的还原势较低,氧化还原能力不平衡,且其带隙稍小,低于其光分解水的效率。于是笔者通过对MoS2进行合金化,发现其分别与MoSe2、MoTe2、WS2合金化之后的单层MoS2的导带底和价带顶的电位相对于水的氧化势和还原势变得较为均衡,但带隙变化很小。为了拉大带隙,对不同合金化的单层MoS2材料进行应力的改变,发现在拉伸情况下单层MoS2及其合金体系的CBM带边位置下移,加剧了氧化和还原的不平衡;而在压力的情况下发现,进行合金化的材料带隙变大,同时CBM带边位置上移,极大地改善了本征MoS2的光催化制氢能力。之后对它们的能带结构和态密度进行了分析,发现单层MoS2进行合金化以及施加应力后,其带边位置的改变,合金元素原子的直接贡献较小,而且合金这种方式与掺杂方式引入孤立掺杂能级进而改变带边位置不同,合金化后的带边位置是整体移动,改善能带结构的同时,没有引入新的复合中心。因此,单层MoS2可以通过与MoSe2、MoTe2、WS2进行合金化,并施加压应力两种手段,拉大其带隙、提高CBM带边位置,进而提高光催化分解水的效率。这对于单层MoS2光催化分解水的实验研究有指导意义。
猜你喜欢
分子催化(2022年1期)2022-11-02
科学导报(2022年43期)2022-07-23
太阳能(2022年4期)2022-05-05
商品与质量(2021年43期)2022-01-18
小天使·聪聪画刊(2021年2期)2021-09-10
建材发展导向(2021年14期)2021-08-23
汽车零部件(2020年10期)2020-11-09
汉语世界(The World of Chinese)(2019年6期)2019-09-10
电子制作(2019年15期)2019-08-27
表面工程与再制造(2019年6期)2019-08-24
