
4,4’-二硝基均二苯脲-d8 的合成
2020-04-22 03:37梁大伟
化工技术与开发 2020年4期
梁大伟
(雅安职业技术学院 药学与检验学院,四川 雅安 625000)
尼卡巴嗪又名球虫净,是由4,4’-二硝基均二苯脲(DNC)与2-羟基-4,6-二甲基嘧啶(HDP)组成的复方药物,作为家禽球虫病的防治兽药使用已经有半个世纪左右,其主要药效组分为4,4’-二硝基均二苯脲。国外许多国家都严格规定了尼卡巴嗪在鸡肉制品中的残留标示物及最高残留限量。2002年,我国农业部将尼卡巴嗪列入了兽药残留检测方法研究优先清单[1];2013 年,食品安全国家标准关于动物性食品中尼卡巴嗪残留标志物残留量测定的规范出台[2]。为了加强兽药残留监控,保障动物源性食品安全,农业部将鸡肉制品中的尼卡巴嗪及代谢物的残留检测列入监控计划目录,成为相关食品中兽药残留检测的必检物质[3]。尼卡巴嗪中的2-羟基-4, 6-二甲基嘧啶在动物体内可迅速通过尿液排除体外,而抗球虫活性成分4,4’-二硝基均二苯脲则会较缓慢地通过粪便排泄,因此目前国外对尼卡巴嗪残留的研究,主要集中在4,4’-二硝基均二苯脲的残留测定上[4]。
稳定同位素无放射性,物性相对较稳定,将其标记到药物的相关位置,可采用液质联用(LC-MS)或气质联用(GC-MS)进行检测,具有更高的灵敏性。近年来,稳定同位素标记药物被广泛应用于兽药残留检测的研究中[5]。作为检测内标物的稳定同位素标记的尼卡巴嗪及其代谢产品具有较高的市场需求,但目前该类稳定同位素标记产品主要以进口为主,国产化较少,价格昂贵,严重制约了我国在食品安全监控领域的技术进步。为了促进该技术在我国的发展,本文拟对尼卡巴嗪活性组成4,4’-二硝基均二苯脲的氘代标记产品进行合成开发。
目前,关于4,4’-二硝基均二苯脲的合成工艺主要有以下一些[6]:1)以对硝基苯胺甲酸乙酯和对硝基苯胺为原料,高温(近150℃)下经酯的芳胺解制得;2)对硝基苯胺与光气反应合成得到;3)对硝基苯胺与尿素经高温(近200℃)缩合而成;4)苯胺先与尿素缩合,然后经硝化反应合成。上述反应均存在反应温度较高、有不易控制的危化品中间体等缺点。鉴于4,4’-二硝基均二苯脲中2 个苯环上有氘的引入,可避免发生氘-氢交换,影响同位素丰度,因此笔者设计以苯-d6为起始原料,经过溴代得到溴苯-d5,然后经过对位硝化[7],最后在钯的催化下与尿素偶联,合成目标化合物4,4’-二硝基均二苯脲-d8[8]。具体过程如图1 所示。
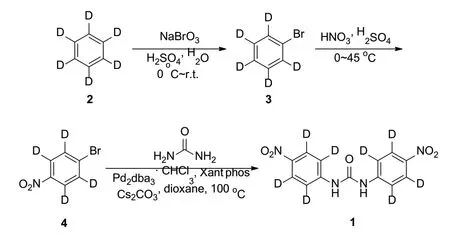
图1 4,4’-二硝基均二苯脲-d8 的合成Fig.1 The synthesis of 4,4’-dinitrocarbanilide-d8
1 实验部分
1.1 仪器与试剂
X-5 型显微熔点测定仪,ARX-400 型核磁共振仪(CDCl3为溶剂,TMS 为内标),6120 Quadruple型LC/MS(ESI 源),Waters 2695 型HPLC(Zorbax Eclipse XDB-C18 column,5 µm,4.6mm×150 mm)。
苯-d6(C6D6)、重水(D2O)(均为外购,化学纯度与氘代丰度都大于99%);尿素、三(二亚苄基丙酮)二钯-氯仿加合物(Pd2dba3-CHCl3)、4,5-双二苯基膦-9,9-二甲基氧杂蒽(Xantphos)、碳酸铯(Cs2CO3)(均为分析纯);1,4-二氧六环(CaH2除水)。其他常规溶剂均为分析纯,未经过进一步处理,直接使用。
1.2 合成方法
1.2.1 溴苯- d5(3)的合成
将浓硫酸(14mL)缓慢加入到50mL 的水中,冰浴冷却10min,然后加入苯-d5(2)(6.6mL,81mmol),分2 批 每 次 间 隔30min 加 入 溴 酸 钠(12.5g,82.5mmol)。添加完毕,继续在冰浴条件下搅拌30min。移去冰浴,升至室温,继续搅拌反应24h。反应完毕,将反应液倒入冰水中,用正戊烷萃取(3×20mL),合并有机相,依次用饱和碳酸氢钠和食盐水洗涤,无水硫酸镁干燥,减压浓缩得黄色液体粗品。常压蒸馏,收集150℃馏分,得10.3 g 淡黄色液体溴苯-d5(3),收率83%。ESI-MS (m/z):163.0 (M+H)+。IR (KBr),ν/cm-1:3063,1580,475,1455。
1.2.2 对硝基溴苯-d4(4)的合成
冰浴条件下,将浓硝酸(4.7mL)与浓硫酸(4.7mL)混合搅拌降温30min,向其中滴加溴苯-d5(3)(5.3g,33mmol),体系中逐渐产生大量的淡黄色固体,20min内滴加完毕。移去冰浴,升温至45℃搅拌反应20min,然后再用冰浴降温,加入30mL 冰水,搅拌10min,抽滤得淡黄色固体粗品。用50mL 乙醇重结晶,除去邻硝基溴苯-d4,得3.0g 白色晶体对硝基溴苯-d4(4),收率45%,m.p.:120~122 ℃(文 献 值[9]为123~125 ℃)。ESI-MS (m/z):207.0 (M+H)+。IR (KBr),ν/cm-1:3105,1632,1621,1470,1400,1370,1350,1323。
1.2.3 4,4’-二硝基均二苯脲-d8(1)的合成
将 对 硝 基 溴 苯-d4(1.0g,4.8mmol)与 尿 素(0.19g,3.0mmol)加入干燥的1,4 二氧六环(50mL)中,常温搅拌,然后分别向上述溶液中加入三(二亚苄基丙酮)二钯-氯仿加合物(0.15g,0.14mmol)、4,5-双 二 苯 基 膦-9,9-二 甲 基 氧 杂 蒽(0.12g,0.2mmol)、碳酸铯(2.2g,6.7mmol),升温回流反应,TLC 监测(乙酸乙酯∶石油醚=1∶10)至反应完毕。冷却至室温,过滤除去不溶物,加入适量水,乙酸乙酯萃取(3×30mL)。合并有机相,饱和食盐水洗涤2 次,无水硫酸镁干燥,减压浓缩得0.58 g 黄色粗品,硅胶柱层析(乙酸乙酯∶石油醚=1∶100),得0.45g黄色固体4,4’-二硝基均二苯脲-d8(1),收率为60%,m.p.>250℃(文献值为310~314℃),纯度为98.6%。HPLC 色谱条件:色谱柱采用Zorbax Eclipse XDB-C18 column,5µm,4.6mm×150mm,流动相为乙腈-水(50∶50),流速为1mL·min-1,柱温为25℃,进样量为20μL,同位素丰度>98%。ESI-MS (m/z):309.1(M-H)-。IR (KBr),ν/cm-1:3340,3110,2230,1635,1590,1455,1410,1340。13CNMR (100 MHz,CDCl3):δ/×10-6:153.3,145.7,143.2,123.3,120.1。
2 结果与讨论
在本合成方法中,第3 步反应中,采用三(二亚苄基丙酮)二钯-氯仿加合物(Pd2dba3-CHCl3)为偶联催化剂,4,5-双二苯基膦-9,9-二甲基氧杂蒽(Xantphos)为配体,碳酸铯(Cs2CO3)为缚酸剂,是成功合成目标化合物4,4’-二硝基均二苯脲-d8的关键。在该关键步骤中,为了提高氘代原料的利用率,降低成本,以尿素过量25%的投料比(对硝基溴苯-d4∶尿素=1.6∶1)进行反应,探究催化剂、配体与缚酸剂的配比对产物收率的影响,结果见表1。以对硝基溴苯-d4的投料量1mmol 为基准,催化剂Pd2dba3-CHCl3为0.03 mmol、配体Xantphos 为0.045 mmol、缚酸剂Cs2CO3为1.4 mmol 时,收率最优。
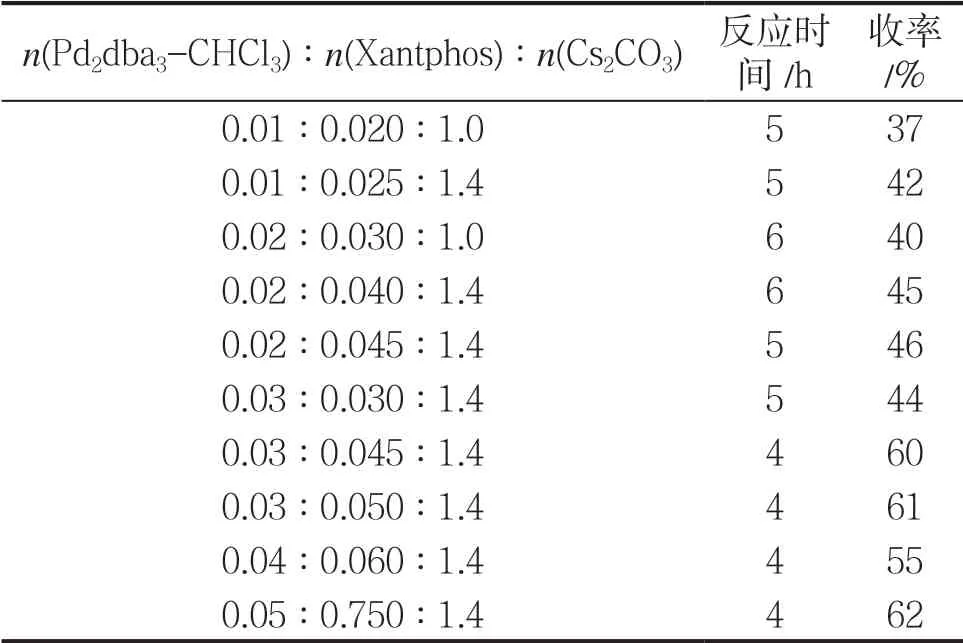
表1 4,4’-二硝基均二苯脲-d8 的合成反应条件优化Tab.1 Optimization of the synthetic conditions of 4,4’-dinitrocarbanilide-d8
3 结论
本文以苯-d6为原料,经溴代、硝基化、钯催化芳基化偶联等反应,以22%的总收率高效合成了4,4’-二硝基均二苯脲-d8。本方法原料易得,过程反应条件易于控制,化学纯度为98.6%,同位素丰度大于98%,可作为家禽类肉制品兽药残留检测用的稳定同位素标记内标药物。
猜你喜欢
化工管理(2021年7期)2021-05-13
鸭绿江(2018年12期)2018-12-28
分析化学(2017年5期)2017-06-21
同位素(2014年3期)2014-06-13
同位素(2014年2期)2014-04-16
同位素(2014年2期)2014-04-16
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
现代计算机(2009年3期)2009-12-21
