
基于生存分析的生物信息学方法筛选乳腺癌枢纽基因和关键通路
2020-04-15 03:17:38刘春良孙海鹏刘云霞
上海交通大学学报(医学版) 2020年3期
陈 思,刘春良,赵 倩,孙海鹏,刘云霞
上海交通大学基础医学院病理生理学系,细胞分化与凋亡教育部重点实验室,上海200025
乳腺癌是最为常见的恶性肿瘤之一,也是女性中发病率最高的恶性肿瘤[1]。根据雌激素受体(estrogen receptor,ER)、孕激素受体(progesterone receptor,PR)和人类表皮生长因子受体2(human epidermal growth factor receptor-2,HER2)的表达情况,可以将乳腺癌分为4种亚型(Luminal A、Luminal B、HER2阳性和三阴乳腺癌)。根据不同乳腺癌亚型的生物学特征和临床病理分期制定相应的个体化治疗策略,可以使乳腺癌5年生存率达90%以上[2]。然而,乳腺癌的复发和转移仍旧是一大难题。除此之外,一些乳腺癌亚型,例如三阴乳腺癌,由于缺乏有效的治疗靶点,一直以来是临床治疗的一个瓶颈。因此,明确乳腺癌发生发展相关的关键基因和通路,有助于认识乳腺癌潜在的发病机制,或将为临床寻找更多的诊断和治疗靶点提供参考。
自基因芯片技术和高通量测序技术问世以来,生物信息学迅速发展,目前已发现了许多疾病的生物学标志物[3-4]。从信息丰富的公共数据库如GEO数据库(Gene Expression Omnibus,基因表达汇编)和TCGA数据库(The Cancer Genome Atlas,癌症基因组图谱)中可获得基因表达数据,通过生物信息学方法,对数据库中的基因表达数据进行聚类分析、统计分析、通路分析和可视化作图等,能够预测基因的功能以及基因间的相互作用,了解疾病基因层面的发病机制,发现潜在的生物学标志物,从而为疾病的分子靶向药物研发和精准治疗提供理论依据。本研究将乳腺癌基因表达数据与临床生存分析相结合,以筛选枢纽基因和关键信号通路;基于生存期筛选出来的基因可能更具有临床意义,或能为乳腺癌的诊断和治疗提供新的思路。
1 材料与方法
1.1 数据采集
为了获取乳腺癌的基因表达数据集,本研究在GEO数据库下载了3个乳腺癌数据集(http://www.ncbi.nlm.nih.gov/geo/),分别为GSE54002、GSE29431和GSE61304,这3个数据集都是基于GPL570平台。GSE54002包含417例乳腺癌样本和16例正常样本。GSE29431包含了54例乳腺癌样本和12例正常样本:54例乳腺癌样本中有15例HER2免疫组织化学评分为3+,且伴有HER2基因扩增;26例评分为2+,其中13例伴有HER2基因扩增,13例不伴有HER2基因扩增;13例评分为0/1+,且不伴有HER2基因扩增。GSE61304包含了58例乳腺癌样本和4例正常样本:58例乳腺癌样本中18例为ER+PR+,19例为ER-PR-,4例为ER+PR-,1例为ER-PR+,其他16例样本未说明。
1.2 筛选差异表达基因
在Rstudio软件中(版本3.4.0)加载并下载Bioconductor网站上软件包来分析上述3个乳腺癌数据集。首先使用affy包导入CEL文件,使用simpleaffy包评估微阵列数据质量[5],gcrma包中的RMA算法预处理原始数据[6],genefilter包过滤非特异性结合的探针和数据质量低的探针,limma包进行差异基因表达的统计学验证[7]。基因表达变化倍数的对数值的绝对值(|log2FC|) >1且P<0.05认定为差异表达基因。最后,为了提高差异表达基因的稳健性,使用Funrich软件(版本3.1.3)获得3个数据集中均上调或下调的基因,用于下一步的分析。
1.3 筛选与总体生存期相关的差异表达基因
为了分析差异表达基因对乳腺癌患者总体生存期的影响,在Kaplan-Meier plotter数据库(www.kmplot.com)中根据基因的中位表达值将患者样本分为高表达组和低表达组。使用默认参数,计算每个基因高表达组和低表达组的中位生存期;若log-rank P<0.05,则该基因被视为与总体生存期相关的差异表达基因。
1.4 分析与总体生存期相关的差异表达基因的功能
基因本体数据库(Gene Ontology,GO)富集分析和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genome,KEGG)通路富集分析被广泛用于识别基因的功能和通路。本研究使用Rstudio软件中clustProfiler包对与乳腺癌患者总体生存期相关的差异表达基因进行GO分析和KEGG分析,P<0.05认为具有统计学意义[8]。
1.5 蛋白 - 蛋白相互作用网络构建及筛选枢纽基因
蛋白与蛋白相互作用在调节生物学过程中起着至关重要的作用。这种关系可以通过蛋白 - 蛋白相互作用(proteinprotein interaction,PPI)网络表示,每个节点代表一个蛋白,边代表蛋白质之间的相互作用。紧密相连的区域可以作为富集功能群。STRING数据库(https://string-db.org/)包含了丰富的蛋白质之间相互作用的信息[9]。为了评估与总体生存期相关的差异表达基因之间的相互关系,将差异表达基因列表导入STRING数据库,并设定信度为0.4;接着将PPI表格数据导入Cytoscape软件中构建PPI网络,使用软件中的插入式分子复合物检测(MCODE评分>4分,节点数>5个)筛选出PPI网络中的枢纽模块,最后通过CytoHubba插件计算网络中每一个基因的最大团中心性(maximal clique centrality,MCC)分数,将得分前10的基因作为枢纽基因[10-12]。
1.6 枢纽基因验证
1.6.1 数据库验证枢纽基因表达 利用Oncomine数据库(https://www.oncomine.org/resource/main.html/)和人类蛋白质图谱(Human Protein Atlas,HPA)数据库(https://www.proteinatlas.org/)对枢纽基因在乳腺癌肿瘤组织和正常组织间的mRNA和蛋白水平的表达进行验证[13]。
1.6.2 RNA抽提及实时荧光定量PCR 人乳腺癌细胞MDA-MB-231和人正常乳腺上皮细胞MCF-10A(购自中国科学院干细胞库)的总RNA抽提按照Invitrogen公司TRIzol试剂盒提供的方法。反转录以1 000 ng RNA为模板,按TaKaRa公司M-MLV Reverse Transcription Kit试剂盒说明配制反转录反应液,进行反转录反应。实时荧光定量PCR(quantitative real-time PCR,qPCR)使用ABI公司7500 Real-Time PCR System试剂盒。每份采用10 μL体系,cDNA模板1 μL,每份样品做3个复孔,求平均值;以18S rRNA为内参,引物序列见表1。

表1 qPCR引物序列Tab 1 Primer sequences for qPCR
1.7 统计学分析
应用OriginPro 2017C和Adobe Illustraor CS6软件进行统计学分析和作图,qPCR数值用x—±s表示,组间比较采用独立样本t检验。P<0.05认为差异具有统计学意义。
2 结果
2.1 筛选差异表达基因
在|log2FC|>1且P<0.05的筛选条件下,从GSE54002中得到差异表达基因3 389个,其中上调基因1 561个,下调基因1 828个;GSE29431中得到差异表达基因3 660个,其中上调基因1 097个,下调基因2 563个;GSE61304中得到差异表达基因1 828个,其中上调基因821个,下调基因1 007个。然后用Funrich软件的Vene图筛选得到了211个共同上调和374个共同下调的差异表达基因,如图1所示。
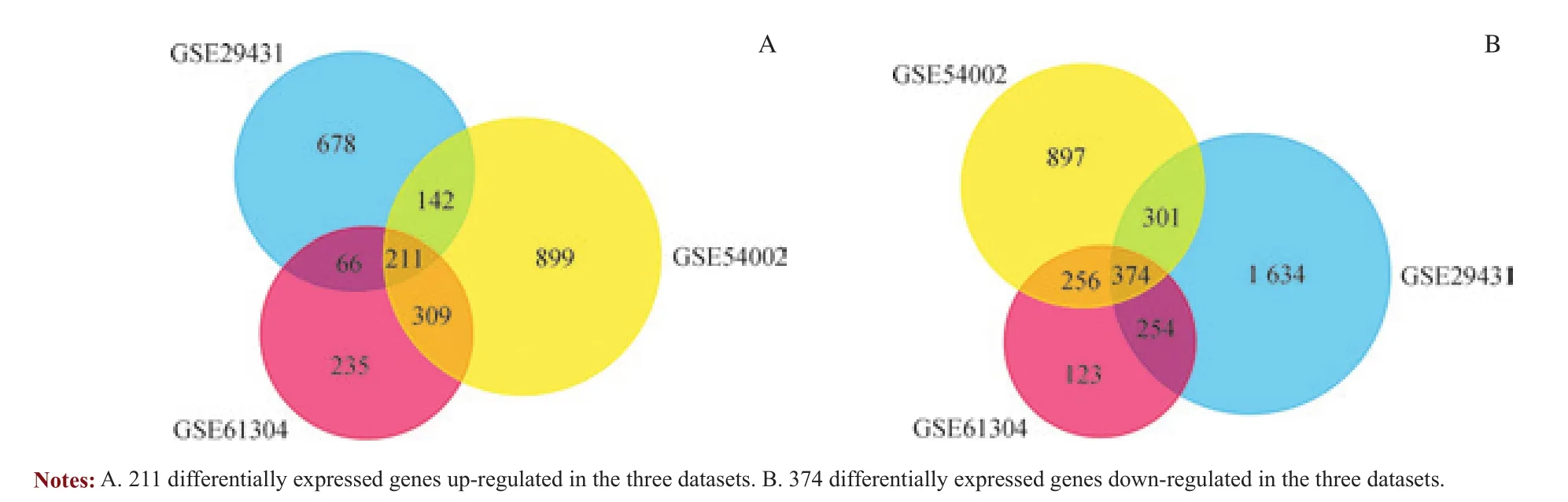
图1 GSE29431、GSE54002和GSE61304中筛选得到的差异表达基因的Venn图Fig 1 Venn diagrams of the differentially expressed genes screened in GSE29431, GSE54002 and GSE61304
2.2 乳腺癌中与总体生存期相关的差异表达基因
为了筛选与总体生存期相关的基因,使用Kaplan-Meier plotter数据库对这585个差异表达基因进行总体生存分析,计算每个基因的高表达组和低表达组的中位生存期和log-rankP值。结果显示,有262个基因的高表达组和低表达组的总体生存期之间的差异有统计学意义。表2列出了与生存相关最显著的前10个差异表达基因。这262个与总体生存期相关的差异表达基因将用于下一步的基因功能分析。
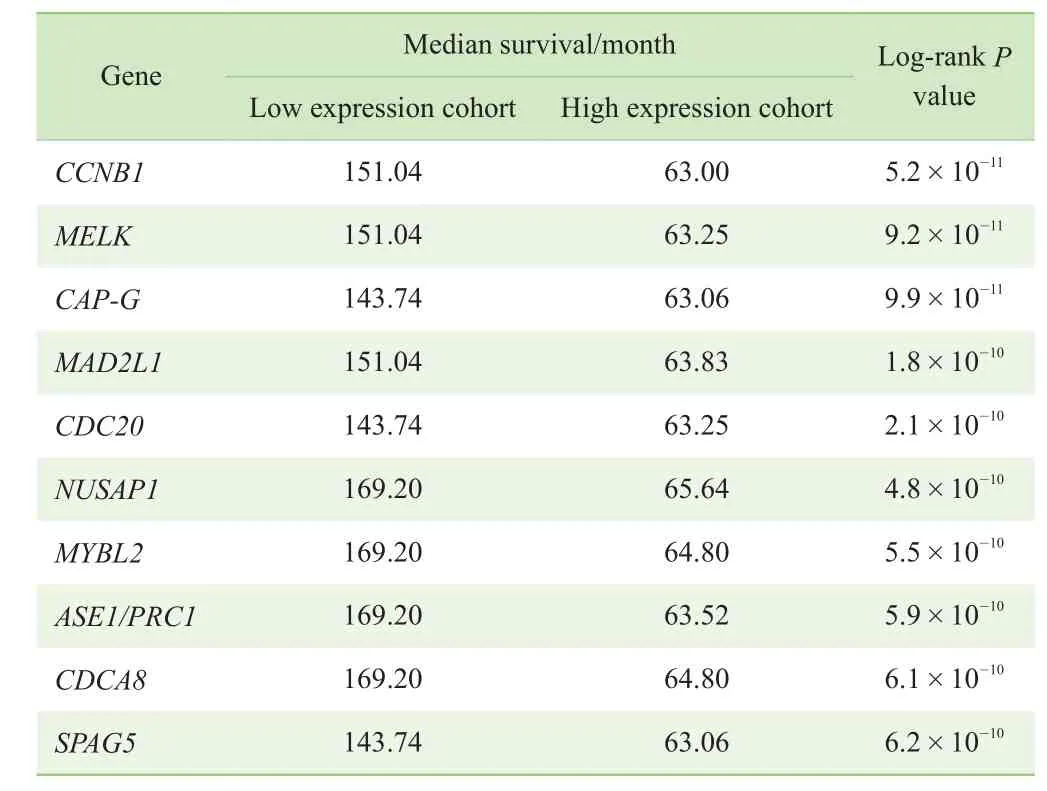
表2 与总体生存期相关最显著的前10个差异表达基因Tab 2 List of the top 10 overall survival-related differentially expressed genes
2.3 乳腺癌中与总体生存期相关的差异表达基因的功能分析
为进一步了解这些基因的功能,利用Rstudio软件中的clusterProfiler包对得到的生存期相关的差异表达基因进行功能分析,P<0.05认为具有统计学意义。GO功能分析结果显示,这些基因的功能主要与细胞分裂(cell division)、核分裂(nuclear division)、细胞器分裂(organelle fission)、细胞周期的调控(regulation of mitotic cell cycle)、负向调控细胞有丝分裂(mitotic nuclear division)以及染色体分离(chromosome segregation)等生物学过程有关(图2A)。KEGG通路分析显示这些基因参与细胞周期(cell cycle)、人T细胞白血病病毒1感染(human T-cell leukemia virus 1 infection)、FoxO信号通路(FoxO signaling pathway)、卵母细胞减数分裂(oocyte meiosis)、细胞衰老(cellular senescence)以及孕酮介导的卵母细胞成熟(progesterone-mediated oocyte maturation)等信号通路(图2B)。
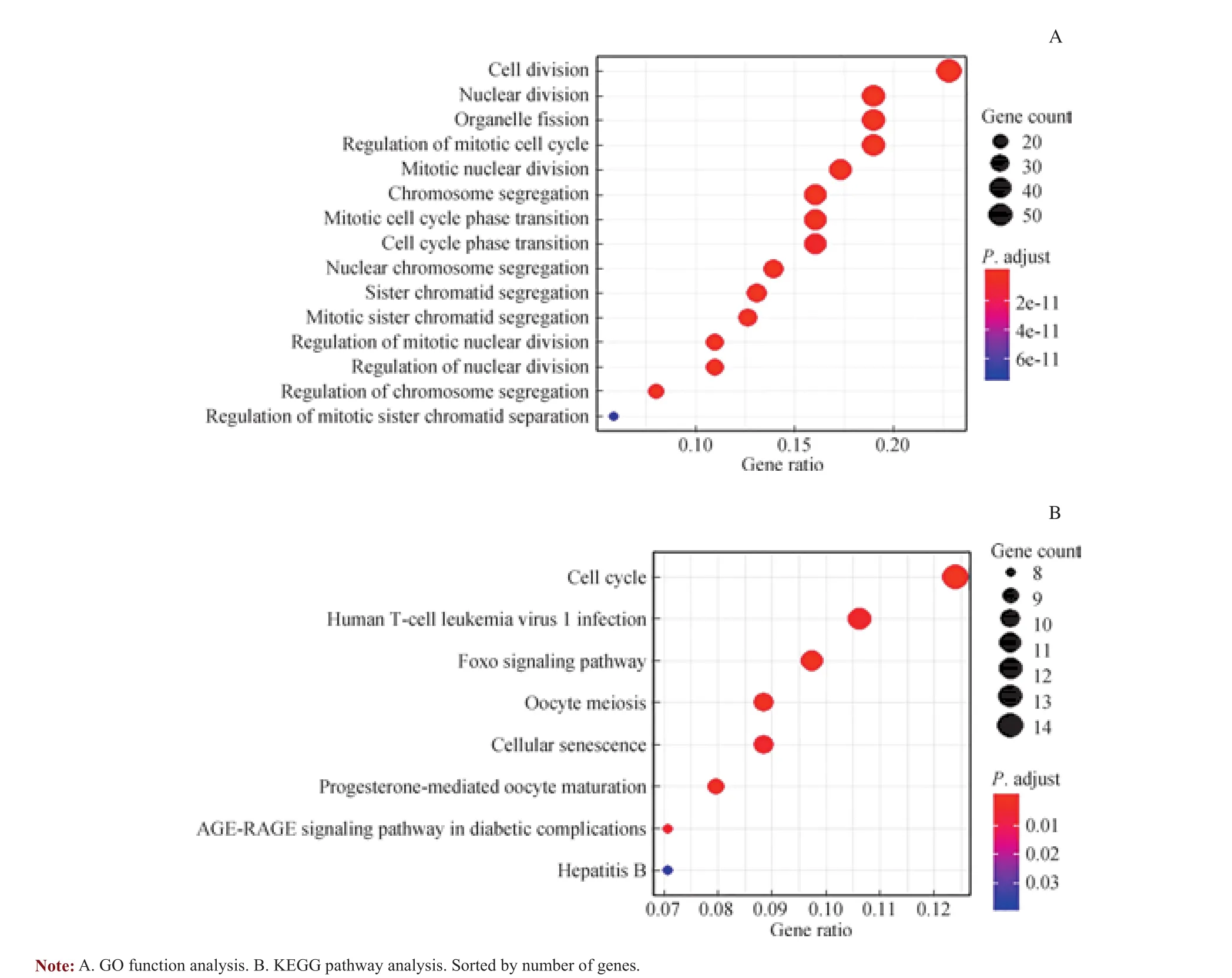
图2 与总体生存期相关的差异表达基因GO功能分析和KEGG通路分析Fig 2 GO function analysis and KEGG pathway analysis of the overall survival-related differentially expressed genes
2.4 PPI网络构建及模块分析
将与总体生存期相关的差异表达基因列表上传至STRING,并设定信度0.4作为判断相互作用是否有意义的标准,构建了PPI网络(图3)。Cytoscape软件根据MCODE评分排序筛选出了3个枢纽模块(图4),并对枢纽模块中的基因进行GO功能分析,结果显示模块1和模块2的基因主要富集在细胞分裂(cell division)、细胞核分裂(nuclear division)和细胞器分裂(organelle fission)等生物学过程,模块3的基因主要集中在血小板脱颗粒(platelet degranulation)、负向调控线粒体细胞色素c的释 放(negative regulation of release of cytochrome c from mitochondria)等生物学过程(表3)。
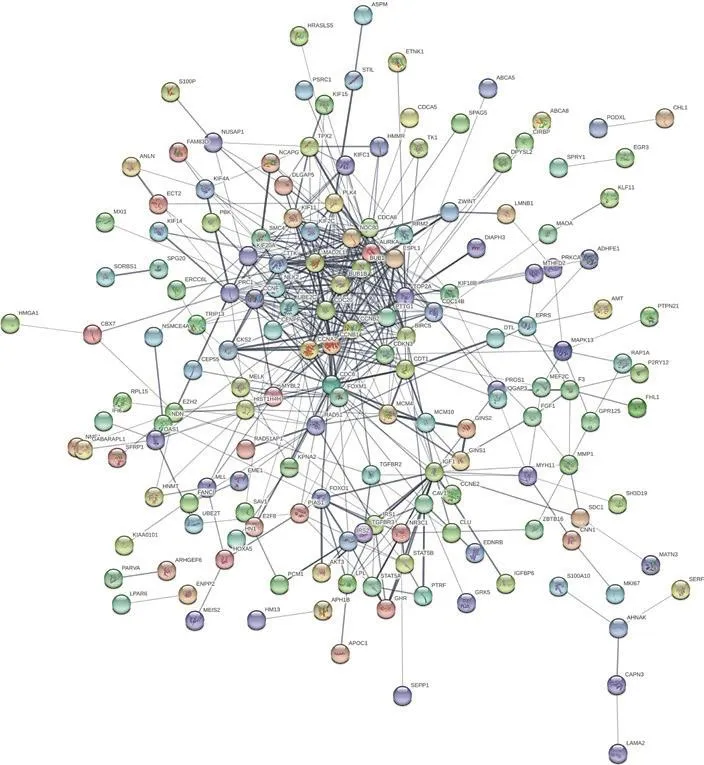
图3 与总体生存期相关的差异表达基因的PPI网络Fig 3 PPI network of overall survival-related differentially expressed genes
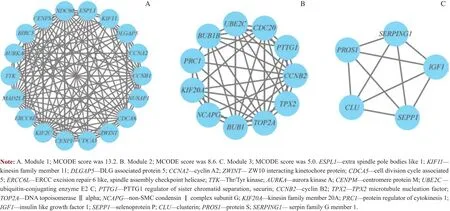
图4 MCODE评分排序筛选出的3个枢纽模块Fig 4 Three modules selected by MCODE scoring sorting

表3 3个模块基因的GO功能分析Tab 3 GO functional analysis of the differentially expressed genes in three modules
2.5 乳腺癌中枢纽基因的鉴定
枢纽基因是一类在生物学过程中发挥至关重要作用的基因,在相关通路中其他非枢纽基因的调控往往要受到这类基因的影响,因此枢纽基因有可能成为乳腺癌的生物学标志物和治疗靶标。使用Cytoscape中的插件Cytohubba,通过MCC法得到了得分前10的枢纽基因,分别是NDC80、BUB1、CDCA8、BUB1B、BIRC5、CCNB1、KIF2C、CENPF、MAD2L1和CDC20(图5)。
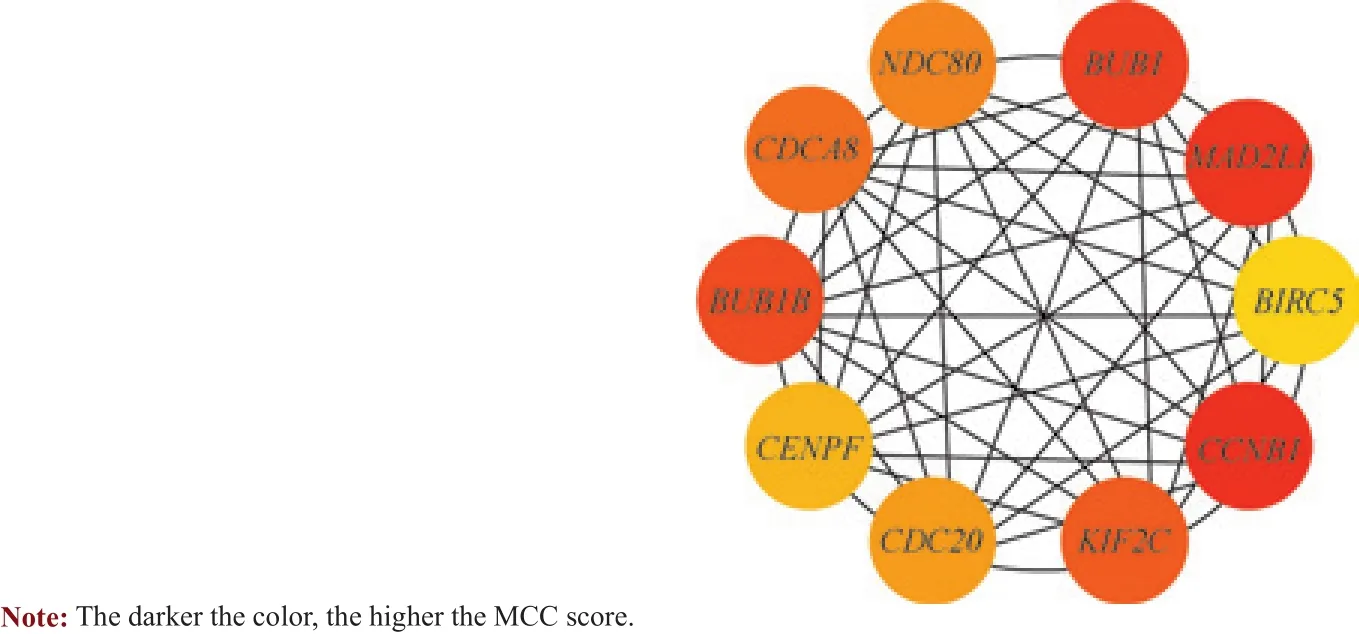
图5 通过MCC法得到的10个枢纽基因及其相互作用Fig 5 Ten hub genes and their interactions by MCC
2.6 枢纽基因的验证
通过Oncomine数据库和HPA数据库对筛选得到的10个枢纽基因进行表达验证。Oncomine数据库结果显示,BIRC5、CDC20、NDC80、CENPF、MAD2L1、CDCA8、KIF2C、BUB1、CCNB1和BUB1B的mRNA水平在乳腺癌组织中明显上调(图6)。HPA数据库免疫组织化学检测结果显示,肿瘤 组 织 中CDCA8、BIRC5、CDC20、CENPF、MAD2L1和CCNB1的蛋白表达水平高于正常乳腺组织,另外4个基因NDC80、KIF2C、BUB1、BUB1B未被HPA数据库收录。以上结果提示,筛选得到的枢纽基因具有较好的稳健性。

图6 通过Oncomine数据库验证枢纽基因转录水平的表达Fig 6 Validation of the expression of hub genes at mRNA level using Oncomine database
此外,通过提取人乳腺癌细胞MDA-MB-231和人正常乳腺上皮细胞MCF-10A的RNA进行反转录和qPCR,验证了这些枢纽基因的表达水平。如图7结果所示,BIRC5、NDC80、MAD2L1、CENPF、CCNB1、BUB1、BUB1B、KIF2C、CDC20和CDCA8在乳腺癌细胞中的表达均高于人正常乳腺上皮细胞;同时,虽然2个枢纽基因MAD2L1与BUB1的表达差异无统计学意义,但乳腺癌细胞中水平仍略高于正常乳腺上皮细胞。以上结果在细胞水平验证了各枢纽基因表达。

图7 qPCR检测乳腺癌细胞枢纽基因的表达Fig 7 qPCR analysis of hub genes in breast cancer cells
3 讨论
本研究利用基于生存分析的生物信息学方法,将基因表达数据与临床生存分析相结合,筛选乳腺癌中的枢纽基因和关键通路。首先,我们从GEO数据库下载了3个乳腺癌组织和癌旁组织的基因表达数据集,筛选出了585个差异表达基因,其中上调的基因有211个,下调的基因有374个。然后用Kaplan-Meier plotter数据库筛选出了262个与乳腺癌患者总体生存期相关的差异表达基因。接着对这262个基因进行了GO功能分析,结果显示这些基因主要与细胞分裂、细胞周期的调控以及染色体分离等生物学过程相关;KEGG通路分析结果表明,这些与总体生存期相关的差异表达基因主要富集在细胞周期、FoxO信号通路和卵母细胞减数分裂等通路上。此外我们发现人类T细胞白血病病毒1感染信号通路在乳腺癌中失调,但目前并无关于人类T细胞白血病病毒1感染信号通路与乳腺癌的报道;由于其在乳腺癌中的分子机制仍不明确,因此需要进一步研究。以上筛选出来的通路可以为乳腺癌发生发展的分子机制研究提供依据。
此外,我们通过构建PPI网络筛选出了10个乳腺癌的枢纽基因,分别为BIRC5、CDC20、NDC80、CENPF、MAD2L1、CDCA8、KIF2C、BUB1、CCNB1和BUB1B,经Oncomine数据库和HPA数据库以及qPCR验证,它们在乳腺癌中均高表达,并与乳腺癌患者较差的生存期相关。通过查阅文献发现这些基因主要参与了细胞有丝分裂染色体的分离和细胞周期的调控。我们还尝试在Oncomine数据库中研究这些枢纽基因的表达是否与乳腺癌分子分型、分期及恶性程度有关;但由于数据库中数据的一些局限,无法直接分析枢纽基因在不同分子分型乳腺癌的表达情况,因此无法确认枢纽基因的表达与分子分型的相关性。但我们发现枢纽基因在ER阴性乳腺癌和PR阴性乳腺癌的表达量分别高于ER阳性乳腺癌和PR阳性乳腺癌的表达量;在HER2阳性或阴性的乳腺癌中,这些基因的表达却没有明显差别(数据未展示),提示枢纽基因的表达可能与分子分型有关。通过分析枢纽基因在不同分期的乳腺癌中的表达情况,我们发现这些基因在ⅠB期的表达量明显低于其他分期的表达量,而BUB1B在Ⅲ期的表达量显著高于其他分期(数据未展示)。MAD2L1、CDC20、CENPF、KIF2C、CCNB1、NDC80、BUB1、CDCA8和KIF2C在各分期的表达量没有明显差异。上述发现说明枢纽基因可能可以作为特定的分子分型和分期治疗的判断依据。
有 研 究 报 道,NDC80、MAD2L1、CDC20、BUB1、BIRC5和CCNB1在乳腺癌的发生发展中发挥了重要功能。NDC80基因编码的NDC80蛋白参与构成微管与动粒连接复合体,为染色体正常分离所必需,在细胞有丝分裂过程中起着至关重要的作用;NDC80的异常表达会造成染色体的异常分离,从而使染色体不稳定,最终导致肿瘤的发生[14]。多项研究[15-16]表明NDC80在多种肿瘤中高表达并参与肿瘤的发生发展。NDC80抑制剂TAI-95可以在体外和体内抑制乳腺癌肿瘤生长,NDC80有望成为乳腺癌的治疗靶点[17]。MAD2L1和CDC20蛋白是纺锤体检查点(spindle assembly checkpoint,SAC)复合体的组成成分,SAC负责确保姐妹染色单体的动粒和纺锤体结合并准确分离到2个子细胞中[18]。研究[19]显示通过shRNA敲除MAD2L1可以抑制乳腺癌细胞的生长和侵袭能力。CDC20的高表达与乳腺癌患者较差的预后相关,且由它介导的BTG3相关核蛋白 (BTG3 associated nuclear protein,SMAR1)降解可以促进乳腺癌细胞迁移和侵袭[20-21]。BUB1是一类丝氨酸/苏氨酸蛋白激酶,在有丝分裂和DNA损伤应答中发挥着重要的功能[22]。Han等[23]发现,敲低BUB1会降低肿瘤干细胞的潜能,BUB1可能成为针对乳腺癌干细胞的治疗靶点。BIRC5编码的蛋白是凋亡抑制(inhibitor of apoptosis,IAP)家族的成员之一,BIRC5具有明显的抑制细胞凋亡的作用。Li等[24]认为BIRC5与乳腺癌患者进展及不良预后相关;研究[25-27]发现,BIRC5是miR-485-5p、ZIC家族成员1(Zic family member 1,ZIC1)和半乳凝素1(galectin-1)的靶点,可能在乳腺癌中发挥重要作用。CCNB1蛋白是调控细胞周期G2/M过渡阶段所必需的;研究[28]表明,CCNB1蛋白与侵袭程度(肿瘤分级、肿瘤体积和淋巴结状态)相关,是乳腺癌的预后因素之一。
目前KIF2C、CENPF、CDCA8和BUB1B这4个枢纽基因与乳腺癌相关的研究还比较少,具体分子机制有待进一步研究。KIF2C和CENPF蛋白参与有丝分裂的染色体分离。研究[29]报道,KIF2C在多种上皮型肿瘤中高表达,并与肿瘤的分级、分期和预后相关。CENPF基因编码一种与着丝粒 - 动粒复合体相关的蛋白,其表达水平在有丝分裂前期增加,并在后期开始时发生蛋白水解[30-31];CENPF的上调与乳腺癌的不良预后相关,可能是有临床意义的治疗靶点[32]。CDCA8是细胞周期分裂相关蛋白家族的一员,此蛋白受细胞周期调控,对染色质诱导的微管稳定和纺锤体形成是必需的;CDCA8的高表达与乳腺癌患者的低生存率和较差预后相关[33]。BUB1B基因编码的蛋白是SAC复合体的重要成员,BUB1B表达增加是恶性程度高的乳腺癌的特征之一[34]。
综上所述,本研究基于与总生存期相关的差异表达基因,筛选了乳腺癌中的枢纽基因和关键通路;基于生存期的枢纽基因可能更具有临床意义,但这些枢纽基因的功能仍需进一步研究。
猜你喜欢
房地产导刊(2020年8期)2020-09-11 07:47:38
商周刊(2019年18期)2019-10-12 08:50:56
当代陕西(2018年12期)2018-08-04 05:49:06
西南国防医药(2016年6期)2016-12-01 06:01:08
癌症进展(2016年11期)2016-03-20 13:16:04
中国病理生理杂志(2015年8期)2015-12-21 12:38:06
哈尔滨医药(2015年6期)2015-12-01 03:58:17
医学研究杂志(2015年3期)2015-06-10 06:41:52
创业家(2015年1期)2015-02-27 07:52:02
中医研究(2014年8期)2014-03-11 20:29:19
