
Primary rhabdomyosarcoma:An extremely rare and aggressive variant of male breast cancer
2020-04-09 08:07linBogdanSatalIoanJungTivadarJrBaraPatriciaSimuIuniusSimuMadalinaVladRitaSzodoraiSimonaGurzu
World Journal of Clinical Cases 2020年19期
Cătălin Bogdan Satală,Ioan Jung,Tivadar Jr Bara,Patricia Simu,Iunius Simu,Madalina Vlad,Rita Szodorai,Simona Gurzu
Cătălin Bogdan Satală,Department of Pathology,Clinical County Emergency Hospital,Tirgu Mures 540139,Romania
Ioan Jung,Rita Szodorai,Department of Pathology,George Emil Palade University of Medicine,Pharmacy,Sciences and Technology,Targu Mures 530149,Romania
Tivadar Jr Bara,Department of Surgery,Clinical County Emergency Hospital,Targu Mures 540136,Romania
Patricia Simu,Iunius Simu,Department of Radiology,George Emil Palade University of Medicine,Pharmacy,Sciences and Technology,Targu Mures 530149,Romania
Madalina Vlad,Simona Gurzu,Department of Pathology,Clinical County Emergency Hospital,Targu Mures 540136,Romania
Simona Gurzu,Department of Pathology,George Emil Palade University of Medicine,Pharmacy,Science and Technology,Targu Mures 530149,Romania
Simona Gurzu,Research Center(CCAMF),University of Medicine,Pharmacy,Sciences and Technology,Targu Mures 540139,Romania
Abstract BACKGROUND Rhabdomyosarcoma(RMS)of the breast,a mesenchymal neoplasm with skeletal muscle differentiation,is an extremely rare tumour in males,with less than 30 cases published in English-language literature.We report on the first case of a male breast RMS,with an unusual ectomesenchymal/neuroectodermal component.CASE SUMMARY A 55-year-old,previously healthy male,underwent a radical left mastectomy for an ulcerated tumour mass,occupying the breast and left anterior thoracic wall.The biopsy specimen indicated the presence of a tumour with neural origins,namely a peripheral neuroectodermal tumour(PNET).The surgical specimens identified two components.The rhabdomyosarcomatous component(over 70%)was represented by large pleomorphic cells with positivity for desmin,sarcomeric actin and myogenin.The PNET-like ectomesenchymal component,which was admixed with the RMS cells,and was also revealed during the preoperative biopsy,consisted of small cells which expressed neurofilament,neuron specific enolase and CD99.The microscopic examination,along with the immunohistochemical profile,allowed the diagnosis of an RMS,with unusual ectomesenchymal differentiation.The patient refused the postoperative oncologic therapy and died three months after surgery.CONCLUSION In patients with RMS of the breast,the PNET-like ectomesenchymal component increases the diagnosis difficulty,especially in biopsy specimens.This differentiation can be immunohistochemically proven and might highlight the possible development of high-grade sarcoma of the breast from remnants of the embryological ectodermal layer.
Key Words:Mammary gland;Rhabdomyosarcoma;Ectomesenchymoma;Male;Case report;CD99
INTRODUCTION
Rhabdomyosarcoma(RMS)is a rare soft tissue tumour which represents less than 1%of all mesenchymal tumours.It mostly occurs in children and young adults between the ages of 15 and 24 years[1-3].The most common histological variant,the alveolar subtype is found in children and the pleomorphic subtype predominates in older adults[3,4].
The majority of RMS cases affect the soft tissue of the head and neck(35%-40% of cases),followed by the genitourinary system(25%),trunk and extremities(with 20%)[3].RMSs which occur in the mammary region are exceedingly rare,with less than 30 cases reported up to February 2020[2].
Independently by localization,the presence of a second phenotype within the RMS,with both rhabdomyoblasts and cells with ectodermic differentiation,defines malignant ectomesenchymoma(MEM)[5].The origin of the second population is thought to be the migratory neural crest cells,embryologically derived from ectoderm,the outer layer of the embryo[5,6].Limited data have been published regarding this dual tumour,with fewer than 20 cases having ever been reported in English-language journals[5],however,none of these were located in the mammary region.
In this paper,we present the case of a male patient,diagnosed with mammary gland RMS with ectomesenchymal differentiation,and we emphasize the complex immune profile needed for a positive and differential diagnosis.The signed,informed consent of the patient was obtained before surgery,for both surgical intervention and the publication of data.
CASE PRESENTATION
Chief complaints
A 55-year-old male presented with a painful,ulcerated tumour mass,located on the anterior thoracic wall in the left mammary region(Figure 1).
History of present illness
The patient reported a six month history of pain in the left mammary region,caused by a progressively growing,ulcerated tumour mass,with no other significant symptoms.Three months before presentation in our department,he underwent a biopsy and suspicion of a tumour of neural origin,namely a peripheral neuroectodermal tumour(PNET)was raised,based on the positivity of tumour cells for Neurofilament and neuron specific enolase(NSE)and negativity for Cytokeratin AE1/AE3.
History of past illness
He was an ex-smoker of seven years and a social drinker,with arterial hypertension and ischaemic heart disease,which were controlled by medication.
Personal and family history
No previous illnesses were declared and there was no family history of oncologic diseases.
Physical examination
The physical examination revealed an ulcerated tumour mass,with infiltrative aspect,occupying the left mammary region(Figure 1).No other signs or symptoms were observed.
Laboratory examinations
A mild elevation of serum level of alanine-aminotransferase(55.4 U/L;normal ranges 0-41 U/L),aspartate-aminotransferase(70.0 U/L;normal ranges 0-40 U/L)and gamma-glutamil-transpeptidase(130 U/L;normal ranges 0-71 U/L)was observed,without any other serological modifications.
Imaging examinations
The preoperative CT-scan revealed a 67 mm × 74 mm × 80 mm nodular mass,located on the anterior thoracic wall,laterally from the sternum,with high density(40 UH)and multiple disseminated micro-calcifications.Direct spread in the soft tissues and tumour-induced costal erosions were also documented,along with axillary and subclavicular adenopathies.The third intercostal space was infiltrated,with no involvement of parietal or visceral pleura.
After surgery,the CT-scan showed a 28 mm × 135 mm(antero-posterior/Laterallateral)haematoma affecting the axillary region and intercostal muscles.No mediastinum involvement or suspicion of bone or lung metastases were highlighted.
Gross and histopathological assessment of surgical specimens
Grossing of the left mastectomy specimen showed a 67 mm × 74 mm × 80 mm tumour mass,located on the inner medial quadrant of the mammary gland,which was associated with a 33 mm × 30 mm ulceration of the skin(Figure 1).On section,it was observed as a poorly circumscribed,yellow-grey tumour mass,with small haemorrhagic areas,which infiltrated the deep soft tissues.The attached muscular components were also macroscopically involved,as were the rib fragments.
Under the microscope,the predominant component,which represented over 70% of the tumour mass,consisted of clusters of large,intensely pleomorphic(>20 mitoses/high power field),elongated cells,with perivascular proliferation but not tumour emboli.Multinucleated cells were also visible.These large cells were admixed with small cells,with amphophilic and scanty cytoplasm and round to oval,centrally located nuclei.This component proved the formation of Homer-Wright-like rosettes,which were composed of specific circular strands of tumour cells with round-oval nuclei,surrounding fibrillary cores.The tumour exhibited an infiltrative growth pattern,with involvement of the subcutaneous tissue and the deep resection margins(Figure 2).
Immunohistochemical profile of tumour cells
Both tumour populations expressed vimentin and did not display positivity for the epithelial markers,such Cytokeratin AE1/AE3 and Cytokeratin 7(Tables 1 and 2).In the predominant component,namely the large and pleomorphic cells,with a Ki67 proliferation index of over 90%,the rhabdoid differentiation was proved in terms of positivity for desmin,sarcomeric actin and myogenin(Figure 3).The tumour cells were not marked by the neural-(neurofilament,NSE),neuroendocrine-(synaptophysin,chromogranin),mesothelial-(calretinin)or vascular markers(CD31,CD34)and did not show a melanoma immunoprofile(negative for S100 and HMB45)(Table 2).
The second component,composed of small cells,showed a Ki67 index of around 40% and a PNET-like aspect.The immunoprofile was similar to those of cells identified in the biopsy specimens.In contrast with the predominant component,these cells showed focal positivity for neurofilament,NSE and CD99(Figure 4),without rhabdoid differentiation.They did not express desmin,Smooth Muscle Actin(SMA),sarcomeric actin or myogenin(Table 2).
FINAL DIAGNOSIS
Based on the histological examination and immunohistochemical profile,the final diagnosis was high-grade mammary gland pleomorphic RMS with neuroectodermal/ectomesenchymal differentiation and deep positive resection margins.
TREATMENT
Based on the imagistic investigations,a radical mastectomy of the left breast was decided upon.The patient signed the informed consent prior to surgery.Following the correct preoperatory preparation,an elliptical incision was made,centred around the ulcerated tumour mass.As the soft tissues were deeply infiltrated,the detachment of the tumour block was carried out simultaneously with fragments of the directly infiltrated pectoral and intercostal muscles and partial resection of the tumourinvaded anterior costal arches of ribs III-IV.Then,an excision of the axillary lymph nodes was conducted and the surgical specimens were transported to the Department of Pathology for further examination.
OUTCOME AND FOLLOW-UP
One month after surgery,the patient presented in our centre,with local recurrence(Figure 1).As the lung and bone metastases were developed and following explanation of the further therapeutic regimen possibilities,he refused bothpostoperative oncologic treatment and tumour re-excision with surgical reconstruction.

Table 1 Immunohistochemical markers used to confirm the rhabdomyosarcoma with neuroectodermal differentiation
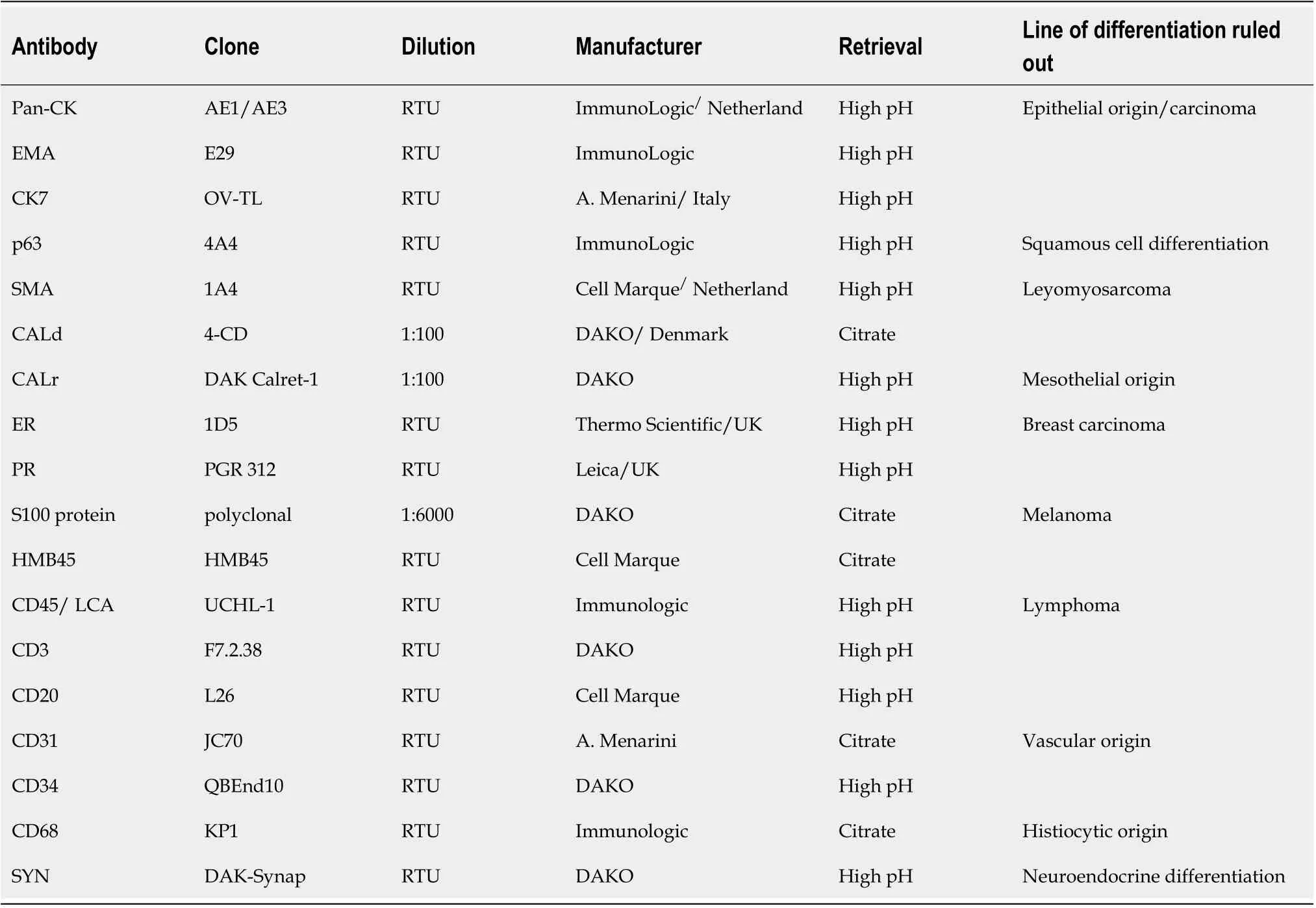
Table 2 Immunohistochemical markers used for differential diagnosis of rhabdomyosarcoma with neuroectodermal differentiation,based on their negativity in the two components
Without any oncological treatment,the patient died three months after the mastectomy.
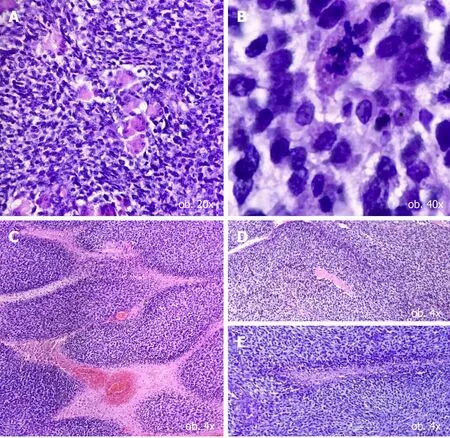
Figure 2 Histological features of a hybrid tumor of the breast.A and B:Major population,some of the cells showing rhabdomyoblastic differentiation(A)and atypical mitoses(B);C-E:Small cell population,demonstrating lobular architecture(C),with formation of rosette-like structures(D and E).
DISCUSSION
To the best of our knowledge,none of the 30 cases of breast RMS,which were previously published in existing English literature[2],were reported in males,despite the fact that the RMSs with other localizations,such as those of the head and neck region,are mostly found in men[6,7].
Although the RMS is a relatively classic malignancy,which can be identified by an experienced pathologist with Haematoxylin-Eosin and only confirmed by the immunohistochemical stains,its rare variants might induce challenges in daily practice.The most common histologic variants of RMS are the embryonal-,botryoid-,alveolar- and pleomorphic subtype[7,8],the latter also being the predominant component of our case.For the diagnosis of sarcomas with dual components,it is mandatory that the histological examination of tumour cells be completed by a large panel of immunohistochemical markers.It is necessary to rule out carcinomas,melanomas,lymphomas and other tumours with neural origin or neuroendocrine differentiation(Table 2).
As regards the genesis of RMS with dual components,it is known that during embryonic myogenesis[4,9-11],myoblasts become mitotically active,then they fuse to form myotubes,which are large elongated cells with abundant eosinophilic cytoplasm.These large cells form primordium of skeletal muscle fibres and can remain in different stages of development,being the key elements of a further malignant tumour[4,9-11].In our case,within the predominant tumour component,large cells resemble the rhabdomyoblasts,which are formed in the early embryologic stage of myogenesis,with some of them also showing multinucleated aggregates.
Our case is of great interest,especially due to the presence of a second,minor cell population resembling MEM,besides the rhabdomyomatous differentiation.MEM,primarily known as gangliorhabdomyosarcoma,is defined as a mixed malignant tumour,one of the elements of which is the RMS component[8,9].It is included within the group of tumours with neuroectodermal differentiation,together with PNET/Ewing sarcomas.This tumour is an exceptional finding,as most of the previously reported cases were diagnosed in children[8,12,13].The case described in this article was firstly diagnosed as a malignant tumour with neural differentiation,due to the predominance of a PNET-like component in the biopsy specimen.
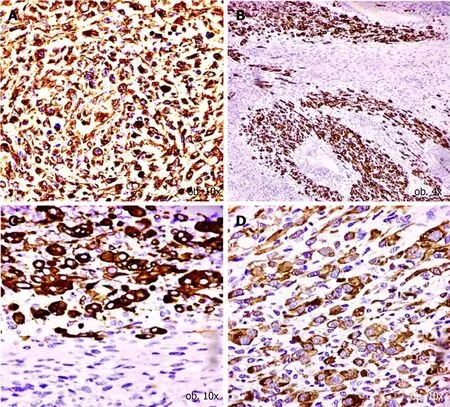
Figure 3 Immunohistochemical profile of major cell population.A:Vimentin diffuse positivity confirms the mesenchymal origin;B-D:The rhabdomyosarcomatous differentiation is confirmed by the positivity for desmin(B),myogenin(C)and sarcomeric actin(D).
The presence of the MEM component,in this case with an RMS phenotype,could be explained by the plasticity of the tumour cells,which might have allowed dedifferentiation.We have not been able to find any study to support this hypothesis,however,the prevalence of this phenomena,mostly in a young population,along with a first diagnosis of pure RMS,may be two plausible arguments which strengthen this theory[14,15].Hence,further studies are needed to confirm/infirm this hypothesis.Another drawback is the lack of a complete microscopic analysis of the tumour mass,as can occur in biopsy specimens.In our case,besides the presence of small CD99 positive cells,admixed within a larger one,another indicator of a second component was the presence of Homer-Wright-like rosettes,composed of circular strands of tumour cells with round-oval nuclei,surrounding fibrillary cores.These are mostly characteristic of tumours with neural or ectodermal origins,as in cases of PNET[13].
Thus,in this case,the diagnosis was RMS with neuroectodermal differentiation.Besides the arguments favouring this diagnosis,there is a further hypothesis related to the specific topography of the tumour.Breast embryogenesis is intimately related to the presence of milk lines[16].These structures emerge from its primordium,known as the mammary ridge,which represents microscopic thickenings of the ectodermal layer of the embryo.These start as microscopic conglomerates of ectodermic cells which migrate from the caudal region proximally,to their final location and to fully develop the mammary glands[13,16].Taking into account these embryogenetic particularities,neuroectodermal differentiation can be present in the sarcomas of the mammary region more frequently than previously thought.
As the diagnosis of MEM is difficult to establish,little is known about its molecular pathways and therapeutic guidelines[3,8,12,13].Radiotherapy with/without chemotherapy can be done before or after surgical excision[17].Kawamotoet al[18]postulated that the evolution of patients with MEM mainly depends on the resectability of the tumor.These fact is also sustained by the study of Freitaset al[19]who demonstrated that incomplete resection of the tumor,such in the present case,is followed by recurrence even though the patients received chemotherapy too.Regarding the chemotherapeutic regimen,its choice primarily depends on the histologic type of the mesenchymal component of MEM.If the chemotherapy targets the mesenchymal RMS component,radiotherapy targets the second,minor population,with neuroectodermal differentiation.The decision of adding or not radiotherapy belongs to the oncologist and should take into account the extent of the neuroectodermal population.In this case,the irradiation dose can go up to 36-45 Gy[17].In cases such as those presented in this paper,therapeutic regimen should be focused on the predominant component,which was the RMS[4,8,9,17].
Figure 4 Immunohistochemical profile of small cell population.A-C:The neuroectodermal origin is confirmed by the positivity for neurofilament(A),neuron specific enolase(B)and CD99(C);D:CD56 positivity proves the interaction between myocytes and neural cells.
We found only two previously published cases of MEM,one by Oppenheimeret al[14]who reported a case of a 17-mo-old boy with MEM of the left wrist,who was disease free,four years after diagnosis.The second case,published by Paikoset al[15]diagnosed a seven-year-old boy with a MEM of the orbit,who had no residual tumour cells after 15 mo of follow-up treatment.This good evolution can be explained by the patient’s age,as tumour cells are more chemosensitive when a patient is younger[14,15],but also by the pure form of these previously reported cases.On the other hand,in our patient,first diagnosis was established three months after occurrence of the tumour(based on the patient’s anamnesis)and another six months passed without any therapy,due to the patient’s refusal,since distant metastases occurred.
CONCLUSION
This case highlights the heterogeneity of sarcomas,which is not as common as in malignant tumours with epithelial origins.On the other hand,although RMS is known to display very aggressive behaviour,almost one year passed from diagnosis to distant metastases.Such cases,diagnosed at an early stage,might present a better prognosis.However,therapeutic guidelines should be more focused on malignancies with dual components.
ACKNOWLEDGEMENTS
The authors thanks to all colleagues involved in the management of this case.
 World Journal of Clinical Cases2020年19期
World Journal of Clinical Cases2020年19期
- World Journal of Clinical Cases的其它文章
- Role of monoclonal antibody drugs in the treatment of COVID-19
- Review of simulation model for education of point-of-care ultrasound using easy-to-make tools
- Liver injury in COVID-19:A minireview
- Transanal minimally invasive surgery vs endoscopic mucosal resection for rectal benign tumors and rectal carcinoids:A retrospective analysis
- Impact of mTOR gene polymorphisms and gene-tea interaction on susceptibility to tuberculosis
- Establishment and validation of a nomogram to predict the risk of ovarian metastasis in gastric cancer:Based on a large cohort