
Gastric plexiform fibromyxoma:A case report
2020-04-08 06:07
World Journal of Clinical Cases 2020年22期
Jin-Yu Pei,Bin Tan,Peng Liu,Guang-Hua Cao,Zu-Sen Wang,Lin-Lin Qu,Department of Hepatopancreatobiliary Surgery,The Affiliated Hospital of Qingdao University,Qingdao 266555,Shandong Province,China
Jin-Yu Pei,Peng Liu,Guang-Hua Cao,Medical College,Qingdao University,Qingdao 266071,Shandong Province,China
Abstract BACKGROUND Plexiform fibromyxoma (PF) is a rare mesenchymal tumor of the stomach. The clinical features of PF frequently include upper abdominal pain,abdominal discomfort,hematemesis,melena,pyloric obstruction and an upper abdominal mass. We herein report a case of PF resected by laparoscopic radical distal gastrectomy plus Roux-en-Y gastrojejunostomy.CASE SUMMARY The patient was admitted to hospital,due to a 1-wk history of an abdominal space-occupying lesion identified during a health examination. He underwent complete resection by laparoscopic radical distal gastrectomy plus Roux-en-Y gastrojejunostomy. During the operation,the tumor was located in the anterior wall of the gastric antrum (approximately 7 cm × 6 cm × 5.5 cm) and did not show evidence of invasion of the serosa. Histology showed that the tumor cells were oval fibroblast-like and spindle-shaped cells,with numerous thin-walled blood vessels and abundant myxoid stroma. Cellular atypia and mitosis were both rare.Immunohistochemistry showed that the tumor cells were immunoreactive for smooth muscle actin,S-100 and CD-10,but were negative for CD-117,CD-34,DOG-1,and ALK. In this case,S-100 was positive and no significant disease was observed during the follow-up period.CONCLUSION The fact that PF is a rare tumor with only a few cases in this region can lead to misdiagnosis of this entity and pose a real diagnostic challenge for general surgeons and pathologists when encountering such patients and differentiating PF from other primary tumors of gastric mesenchymal origin. Our report may help increase awareness of this rare,but important new disease entity.
Key Words:Gastric plexiform fibromyxoma; Plexiform fibromyxoma; Immunohistochemistry; Operation; Mesenchymal tumors of stomach; Stomach; Case report
INTRODUCTION
Gastric plexiform fibromyxoma (GPF) is a rare gastrointestinal mesenchymal tumor that most commonly occurs in the gastric antrum. GPF was first reported by Takahashiet al[1]in 2007,but there have only been a few reports of GPF to date. In these cases,the boundary with surrounding gastric mucosa was unclear,and was plexiform or multinodular; GPF exhibits an invasive growth pattern underneath the mucous membranes and between muscles,and the intercellular substance is full of a mucuslike matrix. Immunohistochemical staining is negative for CD-117,DOG-1,CD-34 and S-100 protein,and Ki-67 staining commonly shows very low proliferation rates that are mostly < 5%. GPF can easily be misdiagnosed as gastrointestinal stromal tumor (GIST)and other mesenchymal tumors,leading to overtreatment. Here,we describe one case of GPF with regard to the current literature and discuss the diagnosis and treatment of the tumor.
CASE PRESENTATION
Chief complaints
A 45-year-old male patient presented due to a 1-wk history of an abdominal spaceoccupying lesion identified during a health examination.
History of present illness
The patient had no signs of jaundice,nausea,vomiting,or fever and was physically healthy.
Personal and family history
The patient had no medical history of malignancy.
Physical examination
The patient’s physical examination was unremarkable.
Laboratory examinations
Laboratory findings were within normal limits,except for slightly higher total bilirubin (31.6 μmol/L; normal range:3-22 μmol/L).
Imaging examinations
Abdominal computed tomography (CT) scan showed a soft tissue mass(approximately 6 cm × 6.4 cm) in the upper right abdominal cavity,and the tumor showed heterogeneous density. Contrast-enhanced abdominal CT scan showed uneven delayed enhancement and a mass located at the gastric antrum of the lesser curvature (Figure 1). Based on the CT scan,GIST was considered highly likely.
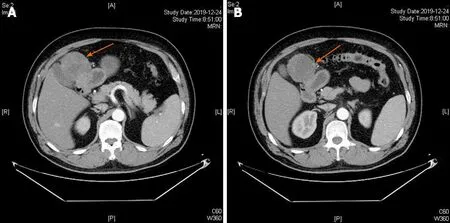
Figure 1 Contrast-enhanced computed tomography axial view revealed a tumor mass with an inhomogeneous enhancement in the arterial phase (A and B).
Clinical diagnosis
The patient was first diagnosed with GIST,which is commonly seen in the General Surgery Department.
Pathology and immunohistochemistry
On gross examination,the size of the submucosal tumor was 7 cm × 5.5 cm × 5 cm,and there was no capsule breakdown. In addition,the cut surface which was gray-red and gray-yellow contained translucent jelly,the quality was slightly tough,and no necrosis of the tumor. Under low-magnification microscopy,the tumor showed a multinodular plexiform growth pattern and was located in the submucosa,and it demonstrated interspersed growth in the smooth muscle and had a clear boundary on the serosal surface (Figure 2A). Under high-magnification microscopy,the tumor cells were oval fibroblast-like and spindle-shaped cells,and there were numerous thin-walled blood vessels and abundant myxoid stroma; cellular atypia and mitosis were both rare(Figure 2B). Immunohistochemically,the spindle cells were positive only for smooth muscle actin (SMA),S-100,and CD-10,but were negative for CD-117,CD-34,DOG-1,and ALK (Figure 3). The Ki-67 labeling index was 1%.
FINAL DIAGNOSIS
According to the results of pathology and immunohistochemistry,the tumor was diagnosed as GPF without lymph node metastasis.
TREATMENT
No tumor was found during physical examination one year previously. Considering the rapid growth of the tumor and the possibility of malignant tumor,the doctor did not perform preoperative biopsy and the patient underwent laparoscopic radical distal gastrectomy plus Roux-en-Y gastrojejunostomy.
OUTCOME AND FOLLOW-UP
After 8 d of treatment,the patient was discharged and had recovered well at 1 mo and 3 mo after surgery.
DISCUSSION
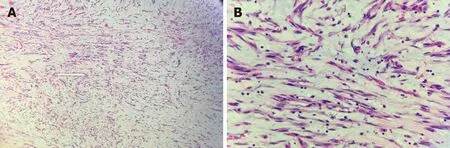
Figure 2 Microscopic examination showed that the tumor exhibited a multinodular plexiform growth pattern. These nodules consisted of blandlooking spindle cells admixed with abundant myxoid or fibromyxoid stroma rich in capillary-sized vessels (hematoxylin and eosin,A:× 100; B:× 400).

Figure 3 Immunohistochemical examinations of the tumor (× 400). Tumor cells showed diffuse cytoplasmic positivity for smooth muscle actin (A) and S-100 (B),but were negative for CD117 (C).
Plexus fibromyxoma (PF) is a rare gastric tumor. To date,121 cases have been reported worldwide[2]. Takahashiet al[1]reported this tumor for the first time in 2007 and named it plexiform angiomyxoid myofibroblastic tumor. In 2010,the World Health Organization adopted the opinion of Miettinen and others,and renamed the tumor PF[3]. The onset of PF is more common during adulthood,but this tumor can also occur in children and adolescents,and there is no gender difference. PF is characterized by a unique cluster of spindle-shaped cell growth and a fine capillary network in a fibromyxoid stroma. PF mostly occurs in the gastric antrum and mainly invades the submucosa and muscularis. There have also been reports of PF in the esophagus,gallbladder,duodenum and colon[4-7].
The clinical manifestations of PF are nonspecific,and this tumor often manifests with nonspecific gastrointestinal symptoms,such as upper abdominal pain,discomfort,hematemesis,melena,pyloric obstruction and an upper abdominal mass.A small number of patients have PF that is incidentally discovered during a health examination. Most fibrous myxomas are submucosal tumors that are smooth or concave and have erosions or ulcerations[8]. Endoscopic ultrasound usually shows that the tumor originates from the muscular layer and is unevenly hypoechoic. CT and magnetic resonance imaging (MRI) are still important methods for the diagnosis of GPF,and these imaging modalities are helpful for tumor examination,staging and the development of surgical plans. Unfortunately,there are reports in the medical literature that most GPF are misdiagnosed as GISTs by ultrasonography and even intraoperative rapid pathology[9].
Of 98 cases,the largest tumor diameter was reported to be 0.8-17 cm,with an average of 4.81 ± 3.30 cm and a median of 4.0 cm[2]. On gross examination,the tumor was located under the mucous membrane and was a nodular or lobulated mass. The boundaries were clear,the surface mucosa was smooth and complete,and there may be small ulcers,depressions,and other features. The cut surface was gray-white,and some were translucent. On microscopy,the tumor appeared to grow in multiple sections,to contain numerous thin-walled blood vessels and to have an abundant myxoid stroma. Tumor cells were oval or fusiform,and cellular atypia and mitosis were both rare. Immunohistochemistry showed that tumor cells had some myofibroblast differentiation,and most PFs were SMA,MSA,and vimentin-positive and CD-34,CD-117,S-100 and DOG-1 negative[10-12]. It is important to note that this patient was S-100 positive. The S-100 protein family consists of 25 known members that regulate a range of different cellular processes,including proliferation,differentiation,inflammation,migration,invasion,apoptosis,Ca2+homeostasis and energy metabolism. The significance of S-100 expression in GPF requires further study.
GPF needs to be distinguished from primary tumors of gastric mesenchymal origin.(1) The most common tumors of gastric mesenchymal origin are GISTs. These tumors show nodular growth,and very few show mucinous degeneration[8].GISTs with mucinous degeneration are more difficult to distinguish from GPF[9,13]. GISTs do not have the characteristic morphology of PF; in addition,the tumor cells are positive for CD-117,CD-34 and DOG-1,and a KIT gene mutation can be detected; however,a few cases of mesenchymoma are CD-117-negative and do not have KIT gene mutation,but a platelet-derived growth factor receptor α gene mutation can be detected[14]; (2)Inflammatory myofibroblastic tumors are common in children and adolescents. These tumors are comprised of proliferating spindle-shaped fibroblasts and myofibroblasts arranged in bundles or swirls,with a large number of inflammatory cells in the intercellular space,and the immunohistochemical marker ALK-1 is positive[15]; (3)Gastrointestinal schwannoma occurs predominantly in the stomach and is mainly composed of spindle cells,most of which form peripheral lymphatic sheaths and germinal centers,with chronic inflammatory cell infiltration and focal infiltrative growth at tumor boundaries,and the immunohistochemical marker S-100 is positive[16]; (4) Leiomyoma cells have the characteristics of an eosinophilic cytoplasm and blunt ends of the nucleus; however,PF generally does not show an obvious bundled arrangement of tumor cells,and the cytoplasm is not as eosinophilic as in leiomyoma. The immunophenotype only shows partial smooth muscle differentiation;and (5) The tumor cells of fibromatosis are arranged in long bundles,and there is no plexiform growth feature. The tumor cells may have β-catenin-positive nuclei. At present,it is still difficult to diagnose the disease by clinical symptoms combined with auxiliary examinations,such as endoscopic ultrasound,CT and MRI. Preoperative aspiration biopsy and frozen section helps to increase the diagnostic rate of the disease,thereby reducing inappropriate treatment,such as imatinib chemotherapy,caused by misdiagnosis in the clinic.
At present,the treatment of this disease is still mainly based on surgical resection,and only a few cases of endoscopic resection have been reported[12,17,18]. The literature suggests that the prognosis is good after surgery,and there is no recurrence or metastasis. This patient remains in good health after 3 mo of follow-up. PF is a benign tumor[19]. However,no cases have confirmed that malignant transformation will not occur[20]. Therefore,confirmation that PF is benign requires longitudinal observation and studies with a sufficient number of cases.
CONCLUSION
GPF is a rare gastric tumor,which has attracted increasing clinical attention and occurs mostly in the gastrointestinal tract. Typical clinical presentations are nonspecific gastrointestinal symptoms,or upper gastrointestinal bleeding. Needle aspiration biopsy is recommended for visualizing the microscopic features of GPF with benign cytological traits,and immunohistochemistry is required for diagnosis as well as for exclusion of GIST. Surgical excision is the main treatment.
 World Journal of Clinical Cases2020年22期
World Journal of Clinical Cases2020年22期
- World Journal of Clinical Cases的其它文章
- COVID-19:A review of what radiologists need to know
- Holistic care model of time-sharing management for severe and critical COVID-19 patients
- Bioequivalence of two esomeprazole magnesium enteric-coated formulations in healthy Chinese subjects
- Osteoprotegerin,interleukin and hepatocyte growth factor for prediction of diabetes and hypertension in the third trimester of pregnancy
- High serum lactate dehydrogenase and dyspnea:Positive predictors of adverse outcome in critical COVID-19 patients in Yichang
- Risk factors analysis of prognosis of adult acute severe myocarditis